
Science:完整人类参考基因组改进了对人类遗传变异的分析
来源:生物谷原创 2022-04-04 16:38
2022年4月4日讯/生物谷BIOON/---人类参考基因组的核心应用之一是作为几乎所有人类基因组研究的比较基线。不幸的是,人类参考基因组的许多困难区域几十年来一直没有得到解决,并且受到塌陷区、序列缺
2022年4月4日讯/生物谷BIOON/---人类参考基因组的核心应用之一是作为几乎所有人类基因组研究的比较基线。不幸的是,人类参考基因组的许多困难区域几十年来一直没有得到解决,并且受到塌陷区、序列缺失和其他问题的影响。相对于目前的人类参考基因组GRCh38,人类T2T-CHM13(Telomer-to-Telomere CHM13)基因组填补了所有剩余的空白,将这种基因组序列增加了近200 Mbp(Mbp),校正了数以千计的结构错误,并为科学探索释放了人类基因组中最复杂的区域。
在一项新的研究中,来自端粒到端粒(Telomere-to-Telomere, T2T)联盟的研究人员展示了T2T-CHM13参考基因组是如何在全球不同的队列中普遍改善读取序列映射和变异识别的。这个队列包括来自扩大的1000基因组计划(1KGP)的经过短读测序的所有3202个样本,以及17个经过长读测序的全球不同样本。通过应用最先进的方法来调用单核苷酸变异(SNV)和结构变异(SV),他们记录了T2T-CHM13相对于之前的人类参考基因组序列的优势和局限性,并强调它有望在基因组的技术挑战区域内揭示新的生物学见解。相关研究结果发表在2022年4月1日的Science期刊上,论文标题为“A complete reference genome improves analysis of human genetic variation”。
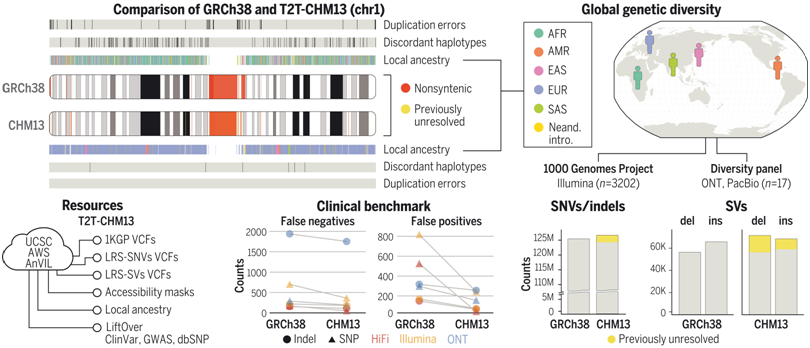
T2T-CHM13的基因组特征和可用资源。图片来自Science, 2022, doi:10.1126/science.abl3533。
在1KGP样本中,与GRCh38相比,这些作者在全基因组范围内利用T2T-CHM13发现了超过100万个额外的高质量变异。在以前未解决的基因组区域,他们在每个样本中发现了几十万个变异---这是进化和生物医学发现的好机会。T2T-CHM13提高了602个三人组之间的孟德尔一致率,并消除了每个样本数以万计的假性SNV,包括将269个具有挑战性的医学相关基因的假阳性率降低了12倍。这些校正在很大程度上是由于在GRCh38中由错误塌陷区域或重复区域引起的长达9 Mbp以上的不准确序列中对70个蛋白编码基因的改进。通过使用T2T-CHM13参考基因组还可以在全基因组范围内更好地了解结构变异,大大改善了序列插入和缺失之间的平衡。最后,通过为T2T-CHM13提供大量的资源(包括1KGP基因型、可访问性掩码和突出的注释数据库),他们的研究工作将促进从目前的人类参考基因组向T2T-CHM13过渡。
综上所述,T2T-CHM13在不同人类谱系样本的变异发现方面取得巨大改进,使得它成功成为人类遗传学的下一个主流参考基因组。因此,T2T-CHM13为构建和研究来自全球不同个体的高质量参考基因组提供了一个模型,比如目前正通过与人类泛基因组参考联盟(Human Pangenome reference Consortium)合作进行的研究。作为一个基础,这项新的研究强调了准确和完整的参考基因组对揭示人类种群多样性的好处。(生物谷 Bioon.com)
参考资料:
Sergey Aganezov et al. A complete reference genome improves analysis of human genetic variation. Science, 2022, doi:10.1126/science.abl3533.
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
