
Molecular Cancer:RIPK3激活诱导癌细胞TRIM28表达下调并增强抗肿瘤微环境
来源:本站原创 2021-08-30 08:45
坏死是一种细胞死亡,可增加肿瘤免疫原性,对治疗免疫荒漠肿瘤有重要意义,因此成为肿瘤免疫治疗的新靶点。
坏死是一种细胞死亡,可增加肿瘤免疫原性,对治疗免疫荒漠肿瘤有重要意义,因此成为肿瘤免疫治疗的新靶点。坏死过程中炎症蛋白的重新合成在促进抗肿瘤免疫反应增强方面显得尤为重要。虽然在细胞死亡过程中由NF-κB介导的晚期转录被认为在这一过程中发挥了作用,但目前还不清楚是什么细胞信号事件启动了这种炎症基因的反式激活。作者采用串联亲和纯化-质谱联用(TAP-MS),结合RNA测序(RNA-Seq)数据集的分析,确定三方基序蛋白28(TRIM28)是一个候选的共抑制因子。综合生物化学和分子生物学技术研究TRIM28在RIPK3激活诱导的转录和免疫调节事件中的作用。用细胞成分估计模块评估所有TCGA肿瘤中RIPK3/TRIM28水平与CD8+T细胞或树突状细胞(DC)的相关性。作者数据表明,癌细胞中RIPK3激活依赖的TRIM28的去抑制导致肿瘤微环境中免疫刺激细胞因子的产生增加,从而有助于强大的细胞毒性抗肿瘤免疫。

图片来源:https://doi.org/10.1186/s12943-021-01399-3
哺乳动物细胞的死亡通过多种机制对不同的应激做出反应;细胞死亡的异常调节导致了各种人类疾病,如神经退行性变、自身免疫性疾病、传染病和癌症。坏死是一种有规律的坏死细胞死亡;其基本的分子机制包括两个受体相互作用蛋白激酶(RIPK1和RIPK3)和混合谱系激酶域样伪激酶(MLKL)。RIPK3的磷酸化是核心坏死途径的一个重要方面,这随后导致磷酸化的MLKL,从而诱导质膜的低聚化和易位,从而导致膜渗透。坏死细胞可能在先天免疫中发挥多种作用,并通过释放内源性危险信号即损伤相关分子模式(DAMPs)塑造随后的适应性免疫。越来越多的证据表明,随着坏死的诱导,细胞因子和趋化因子的重新合成发生在细胞死亡时,然后影响免疫过程。事实上,RIPK1/RIPK3的激活导致炎症趋化因子的上调,从而促进CD8+T细胞免疫应答的交叉启动;肿瘤内趋化因子的存在与细胞毒性CD8+T细胞(CTL)浸润呈正相关,表明坏死信号的激活提供抗肿瘤免疫。
一些研究最近提出了在坏死过程中产生免疫刺激细胞因子的机制。这些报告显示,NF-κB转录和翻译活性参与了RIPK1/RIPK3激活依赖性坏死。当然,肿瘤坏死因子(TNF)介导的坏死是通过持续的NF-κB活化来增强炎症因子基因转录的,这在很大程度上是通过众所周知的机制实现的;然而,RIPK3活化本身是如何导致NF-κB持续活化的还不完全清楚。细胞本身激活细胞因子产生的其他机制也可能存在。
TRIM28是60个TRIM家族蛋白之一,是一种参与基因表达的转录调控因子,部分是通过与Krüppel相关的box抑制域的相互作用介导的,这些域通常存在于转录因子中。TRIM28与异染色质蛋白1(Hp1)一起位于异染色质中,位于TRIM28羧基末端的植物同源结构域手指和溴域招募各种转录共抑制因子,包括核小体重塑去乙酰化酶(NuRD)、组蛋白去乙酰化酶复合体和组蛋白H3赖氨酸9特异性甲基转移酶SETDB1来抑制基因的表达,其中包括核小体重塑去乙酰化酶(NuRD)、组蛋白去乙酰化酶复合体和组蛋白H3赖氨酸9特异性甲基转移酶SETDB1(组蛋白H3赖氨酸9特异性甲基转移酶SETDB1)。TRIM28的辅抑制因子功能最近被证明与各种癌症的发展有关,如非小细胞肺癌、乳腺癌、宫颈癌、结肠癌、胃癌和卵巢癌。然而,TRIM28是如何参与由外部刺激触发的主动转录的,目前尚不清楚。
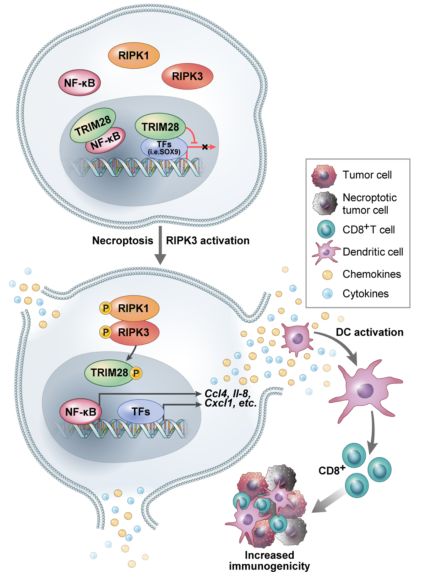
建议的RIPK3/TRIM28激活介导的免疫刺激细胞因子产生示意图
图片来源:https://doi.org/10.1186/s12943-021-01399-3
在这里,作者证明了TRIM28是一种负转录调节因子,当发生坏死的细胞继续从头合成免疫刺激细胞因子时,它本身就是负调控的。TRIM28可以拮抗NF-κB的反式激活,而不依赖于p65染色质的占有率,但RIPK3激活介导的丝氨酸473位TRIM28的磷酸化有助于其去阻抑。此外,RIPK3的激活触发染色质中TRIM28结合事件的显著减少,从而导致SOX9转录因子活性的增加。作者的结果揭示了一种新的坏死介导的转录回路,该回路受RIPK3激活依赖的TRIM28下调的调节,这为促进强大的抗肿瘤免疫提供了一种机制,并有助于肿瘤的免疫原性。(生物谷 Bioon.com)
参考文献
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
