
柳叶刀·肿瘤学|中西对话:再谈FDA对抗肿瘤新药的监管考量
来源: 医药魔方 2022-05-14 08:46
5月初,中国创新药出海的另外两个标志性事件给出了中期答卷,君实生物和和黄医药均收到了FDA的完全回复函(Complete response letter,CRL)。其中,特瑞普利单抗的鼻咽癌上市申请被
5月初,中国创新药出海的另外两个标志性事件给出了中期答卷,君实生物和和黄医药均收到了FDA的完全回复函(Complete response letter,CRL)。其中,特瑞普利单抗的鼻咽癌上市申请被要求进行一项质控流程变更,需要重新提交上市申请;索凡替尼的神经内分泌瘤上市申请则被直接认为是证据不足,需要开展纳入更多美国患者的国际多中心临床试验来支持美国市场的批准。
再往前追溯的话,FDA在今年2月11日召开了一场面向公众的肿瘤药物咨询委员会(ODAC)会议,讨论信迪利单抗NSCLC适应症的美国上市申请问题。最终,ODAC专家组是以14:1的投票结果认为信迪利单抗需要补充额外的临床试验,以证明其在美国患者人群中的适用性,再重新递交上市申请。之后不久,信达的合作伙伴礼来也正式对外宣布收到了FDA发布的完全回复信。
上述3款中国创新药被FDA陆续拒绝批准在国内外引起了巨大的关注,各个媒体也对此做了很多报道。5月初,《柳叶刀肿瘤学》(The Lancet Oncology)杂志同时刊登了两篇分别来自中、西方学者专家的文章,结合信迪利单抗这一案例就“FDA肿瘤新药审批决策”展开了对话讨论。其中,中国学者主要基于进口药审评的多年经验,对基于海外临床数据进行监管决策的考量点与FDA进行了回应,而西方专家则从四个方面举出反例讨论了FDA基于“单一海外国家临床数据”做出决策的矛盾与区别对待之处,让我们一起来看看双方的深度解读。

中方专家观点:中国药监机构针对进口药品的监管经验
自20世纪90年代以来,中国在很长时间内高度依赖进口创新药物来满足国内的医疗需求,监管机构对于新药上市申请做出的监管决策也主要是根据来自美国和欧洲的临床数据。与FDA的监管考虑相似,中国也一直在动态变化的疾病临床需求和药物临床获益的框架内进行获益-风险评估。事实上,随着药物开发的全球化,基于境外单一国家临床数据进行审批决策并不是FDA独有的问题,而是各国监管机构都会面临的共性问题。
首先,未满足临床需求的程度和试验药物临床获益的大小将决定监管机构采取积极还是谨慎的监管决策。当临床获益显著时,药监部门可能会愿意承担中国数据较少甚至缺乏所带来的不确定的风险。举例来说,KEYNOTE-189研究并未纳入中国患者,但基于其在OS和PFS方面的明显获益,同时帕博利珠单抗在另外两项在NSCLC的III期研究(047和042)中均显示出确切的疗效,以及在其他适应症的多项临床试验中对中国人群显示出良好的安全性,NMPA在KEYNOTE-189研究中没有中国数据的情况下也批准了帕博利珠单抗联合化疗用于转移性非鳞状非小细胞肺癌(NSCLC)的一线治疗。
同样的,FDA加速批准了泽布替尼用于复发/难治性套细胞淋巴瘤的治疗,主要根据在中国进行的一项关键性单臂II期研究(BGB-3111-206)数据,并用来自在澳大利亚、美国及其他国家进行的一项B细胞恶性肿瘤I期研究(BGB-3111-AU-003)数据作为补充。笔者认为中美监管机构在这点上是一致的,是基于临床需求和获益做出的积极决策。这一监管评估原则不仅适用于肿瘤领域产品,也适用于非肿瘤领域产品。例如,近期NMPA基于国外的临床数据批准了Paxlovid (Nirmatrelvir-ritonavir)用于COVID-19的治疗。
其次,临床需求和治疗前景的动态变化,很大程度上影响了试验设计和监管标准。例如HER2阳性乳腺癌的二线治疗,随机对照试验(RCT)中的对照组药物随着标准治疗的变化而变化,从化疗到拉帕替尼再到T-DM1。最佳对照组的使用可能会增加试验的外在效度(external validity),从伦理角度保护了受试者的权益。但有数据显示,FDA批准的抗肿瘤药物中,约四分之一使用了次优对照RCT,并非监管降低了标准,更可能的原因是这些试验进行时新的标准治疗方案可能不可及或者还不是当时的标准治疗,不同国家地区可能上市时间不同导致标准治疗不同。中国在2021年11月颁布的肿瘤药物临床开发指南中也强调“对照组应是临床实践中广泛使用的、最佳的标准治疗”。回到信迪利单抗的案例,ORIENT-11试验启动于2018年,当时帕博利珠单抗还未获批该适应症。因此。用化疗作为对照组,在当时的中国临床实践情况下来说是可以被接受的,当然已不再适用于现在的临床实践。
第三,在特定的未满足临床需求和治疗获益的背景下,是否可以将国外数据推到中国患者人群,监管机构还需要谨慎评估种族差异的风险。国际人用药品注册技术协调会(ICH)E5指南是中国审评机构进行种族因素评估的重要参考,在2017年加入ICH前即如此要求。根据种族差异的程度,监管机构可能会在批准前提出不同的要求,包括进行桥接试验、额外的RCT等。由于临床病理学特征和医疗实践的差异,仅根据国外试验数据可能会增加不确定性;因此,可能需要在中国患者人群中进行额外的关键性研究来证明其有效性和安全性。例如,索拉非尼申请治疗晚期肝癌适应症时,考虑到亚洲和西方国家之间肝细胞癌的病因不同,申办方被要求在亚太地区额外进行一项随机对照试验,检验索拉非尼与安慰剂相比的优效性(NCT00492752)。同样,帕博利珠单抗申请用于治疗转移性黑色素瘤适应症时,考虑到不同人种患者中恶性黑色素瘤病理学特征的广泛变异性,申办方被要求在中国开展一项额外的研究(KEYNOTE-151),以证明其在中国患者中的抗肿瘤活性。
第四,根据ICH E17指南的建议,国际多中心临床试验(MRCT)被认为是促进药物同期开发以及探索群体间疗效一致性的有效方法。NMPA认识到MRCT的优势,并一直努力改善中国参与MRCT的监管和临床开发环境。因此,对于进口药品的发展策略,已经从海外药品获批后进行桥接试验,转向在中国获批前加入MRCT。这一策略可以大幅度降低中国药品的滞后问题。从下图来自2005-2021年的中国进口抗肿瘤药数据可以看到这一趋势。
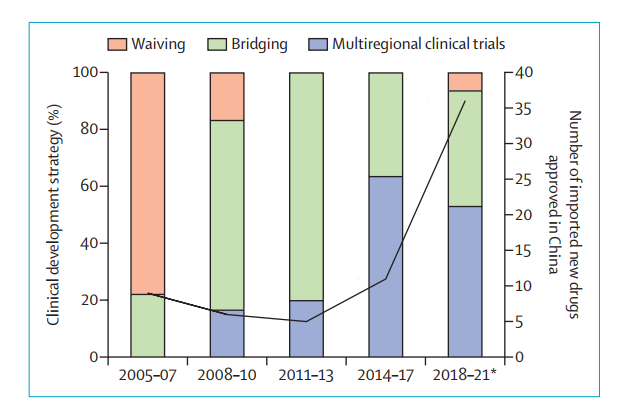
2005-2021年,中国进口抗肿瘤药物临床开发策略(粉色列:表示在中国获批以前,没有在中国人群中进行临床试验;绿色列:表示在中国进行桥接试验;蓝色列:表示中国参与的国际多中心临床试验。数据截至2021年5月31日)
最后,研究数据质量也是监管决策中的关键考量因素。需要指出的是,FDA从《BMJ Opinion》上引用“来自中国80%的临床试验数据是欺诈或不合格的”具有一定误导性。2015年7月22日,CFDA(现NMPA)发布了《关于开展药物临床试验数据自查核查工作的公告》,后续约75%的申请被自愿撤回。随后,NMPA为提高临床数据的完整性、真实性和合规性实施了一系列严厉措施,包括出台处罚政策、发布新版《药物临床试验质量管理规范》、在中国实施ICH E6指南等。因此,近些年在中国进行的临床试验,数据的完整性和质量都得到了极大的改善。自2015年以来,越来越多的来自中国临床研究中心的试验数据通过了FDA的检查,证明了中国的试验数据的质量和可靠性,也是符合FDA标准的。
西方专家观点:FDA在使用单个非本国试验数据方面没有采用统一标准
加利福尼亚大学的David J Benjamin教授等人从研究终点、对照组标准治疗、人群代表性和市场竞争四个方面说明这次FDA的决策没有统一执行标准,没有平等对待所有公司。
首先,对于临床终点是PFS不是OS的这点来说,ODAC认为选择的终点不合理。但是,FDA一直都有使用OS以外的替代终点来批准药物的先例。1992年至2019年间,FDA约有194次使用替代终点批准抗肿瘤药物,基于PFS的常规批准有58次,加速批准有9次。仅在肺癌中就有多种药物是根据缓解率或肿瘤退缩程度获得批准,包括塞瑞替尼、吉非替尼、克唑替尼、奥希替尼、洛拉替尼等。
其次是对照组为化疗不适用于美国的标准治疗的问题,ORIENT-11试验是在FDA批准帕博利珠单抗3天后,也就是2018年8月23日开始招募患者的,但帕博利珠单抗在2019年4月1日才通过NMPA批准。FDA提出对照组不是美国的标准治疗,但是FDA并没有一以贯之的执行该标准。有数据显示,FDA批准的药物中有17%使用了不符合美国标准治疗的对照组。
第三,FDA和ODAC认为ORIENT-11试验不能代表美国患者人群。但是,同样是免疫检查点抑制剂,且临床试验未包括美国患者人群,对照组也不符合美国标准治疗,FDA批准了Cemiplimab用于PD-L1 TPS≥50%晚期NSCLC的治疗。目前,尚没有前瞻性试验设计来比较不同种族之间免疫检查点抑制剂的疗效差异。既往研究显示,纳武利尤单抗和帕博利珠单抗在不同人种或种族之间未显示出任何药代动力学方面的差异。
最后,FDA本身也反复强调欢迎竞争,并在同一领域批准了多个类似的药物,如用于转移性尿路上皮癌的阿替利珠单抗、阿维鲁单抗、度伐利尤单抗、纳武利尤单抗和帕博利珠单抗。然而,在评估信迪利单抗的时候,FDA却表示加剧竞争并不是其监管程序的目标,这一反转也令人困惑。ODAC最终以14:1的投票结果要求信达进行额外的试验。唯一的异议者是美国南加州大学Jorge Nieva教授,他认为信迪利单抗的获批将增加患者的选择和可及性,并降低医疗费用。
总体来说,FDA反对信迪利单抗获批的论点是有说服力的,但是FDA这次的标准和监管决策与之前的声明缺乏一致性。对照组设计问题、替代终点、基于相似数据批准多种药物是抗肿瘤药物监管的常态。FDA可能希望朝着不同的方向发展,但必须谨慎统一执行标准和惯例。药品监管需要良好的信誉,因此,FDA应提供明确的指导,并平等对待所有公司。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
