
Nature子刊:智能与模拟计算双驱动加速PROTAC药物研发
来源:生物世界 2022-09-19 18:07
利用智能与模拟计算双驱动加速PROTAC药物研发,在短短49天内发现了具有高降解活性及高药代性能的新型先导化合物并完成湿实验验证。

自2001年首次对蛋白分解靶向嵌合体(PROTACs)进行概念验证以来,PROTACs已成为通过泛素-蛋白酶体系统选择性降解靶蛋白的革命性工具。PROTACs包含三个部分:一个针对靶蛋白质(POI)的配体(亦称弹头)、一个招募E3泛素连接酶的配体、以及一个连接两个配体的化学连接体。由于这种双功能结构,PROTACs有能力同时结合靶蛋白和E3泛素酶,形成活性三元复合物。因此,PROTACs只需要短暂地与目标蛋白结合就能诱导泛素化和降解。除此之外,PROTACs不需要占据可药用的活性位点这一特性,使利用目标蛋白的所有表面结合位点来调节“不可成药”靶点成为可能。然而,目前PROTACs的设计和优化仍需要基于经验的反复迭代和优化,这种开发策略存在着一定的的局限性。
在PROTACs的开发过程中,最棘手的挑战是如何选择合适的连接基团,从而形成合适的PROTAC活性三元复合物,以发挥降解活性和靶点选择性。由于三元结构的复杂性和动态性,连接基团的设计往往是一个艰巨的挑战。连接基团的长度、组成、柔性、连接位点都会对结果造成巨大的影响。此外,另一个设计挑战则来自于PROTAC分子往往不符合口服药物常见的性质。作为一种多组分分子,其较大的分子量导致其相比于传统小分子有着溶解度差、渗透性差、生物利用度低以及不可预测的Hook效应等问题,从而阻碍了PROTACs的临床转化。因此,如何在有限的条件下理性优化PROTAC分子以克服这些问题,是目前该领域的重大难题。
智能与模拟计算双驱动
PROTAC-RL仅用49天发现新型先导化合物
为了解决这一问题,研究团队提出了一种基于深度生成模型的PROTAC理性设计算法——PROTAC-RL。该模型以一对E3配体和弹头作为输入,输出设计好的连接物,并在强化学习(Reinforcement Learning,RL)的引导下生成具有特定性质的PROTAC分子。具体而言,研究团队首先用Transformer神经网络预训练一个连接子生成模型。然后,在模型训练过程中,为了克服PROTAC训练数据少的问题,该模型先使用大量与PROTACs具有相似化学空间的类PROTAC分子做预训练,再通过真实的PROTACs及增强数据进行模型微调。随后将训练好的模型结合到一个具有经验奖励功能的记忆式强化学习模块中,以产生具有更好药代动力学属性的PROTACs。
作为概念验证,研究团队选择了BRD4作为靶蛋白,并生成了5000余个PROTACs。依托超算能力,研究团队通过高通量的机器学习打分器和分子动力学模拟方法,对这些虚拟分子进行了聚类和筛选。根据合成可及性,研究者最终挑选、合成并实验测试了6个PROTACs,其中3个显示了对BRD4的抑制性活性,1个先导化合物同时显示出对肿瘤细胞系的高抗增殖效力,并在小鼠中表现出良好的药代动力学(图1)。基于星药科技在PROTAC研究方向的长期探索沉淀、中山大学国家超算广州中心大规模并行分子动力学模拟方法以及用于深度学习模型训练的海量GPU算力,该研究整个过程仅耗时49天,证明了超算、深度学习和分子动力学的结合能够促进高效的理性PROTAC设计和优化。
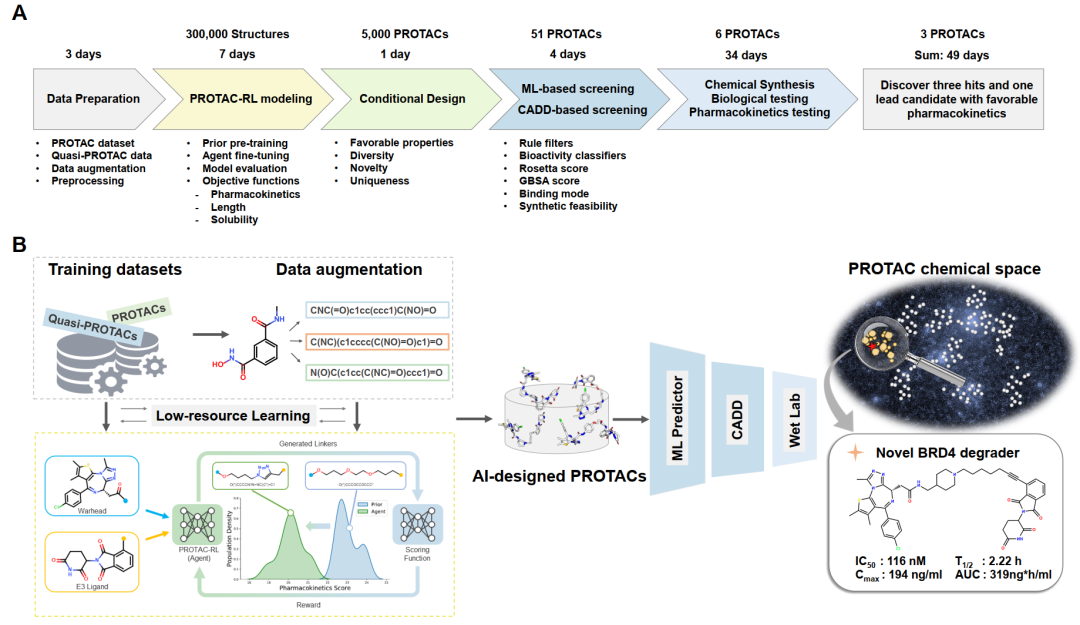
PROTAC-RL模型由两部分组成,基础生成模型Proformer和成药性强化学习模型RL。在评估Proformer的工作中,研究者首先将PROTAC数据集以8:1:1的比例进行切分,分别用于训练、验证和测试。对于弹头和E3配体的每个测试对,生成10个候选PROTACs,并使用复现测试集真实PROTACs的百分比(复现率)来评估性能。研究者将Proformer与其他最先进的片段连接方法进行了比较,包括基于图学习的方法Delinker、基于序列的方法Syntalinker、以及它们在PROTAC训练集上的重训练版本。如图2-A所示,Proformer达到了43.0%的复现率,明显优于现存最好的基线方法。其生成的分子相比于其他方法,也与PROTAC的真实化学空间更加相近(图2-B)。同时,消融实验(图2-C)也再次证明模型设计的合理性。配合强化学习后,PROTAC-RL模型生成的分子打分也远高于其他模型变体(图2-D)。在一个挑选的案例中,PROTAC-RL模型可以随着目标分数设定的不同,生成特定性质的连接基团(图2-E、F)。总的来说,与其他方法相比,PROTAC-RL模型在复现率、有效性、独特性和新颖性等表现上都有着优越的表现。

图2. PROTAC-RL与最新预测方法的比较结果以及消融实验的结果
验证案例:针对BRD4的PROTAC设计及湿实验验证
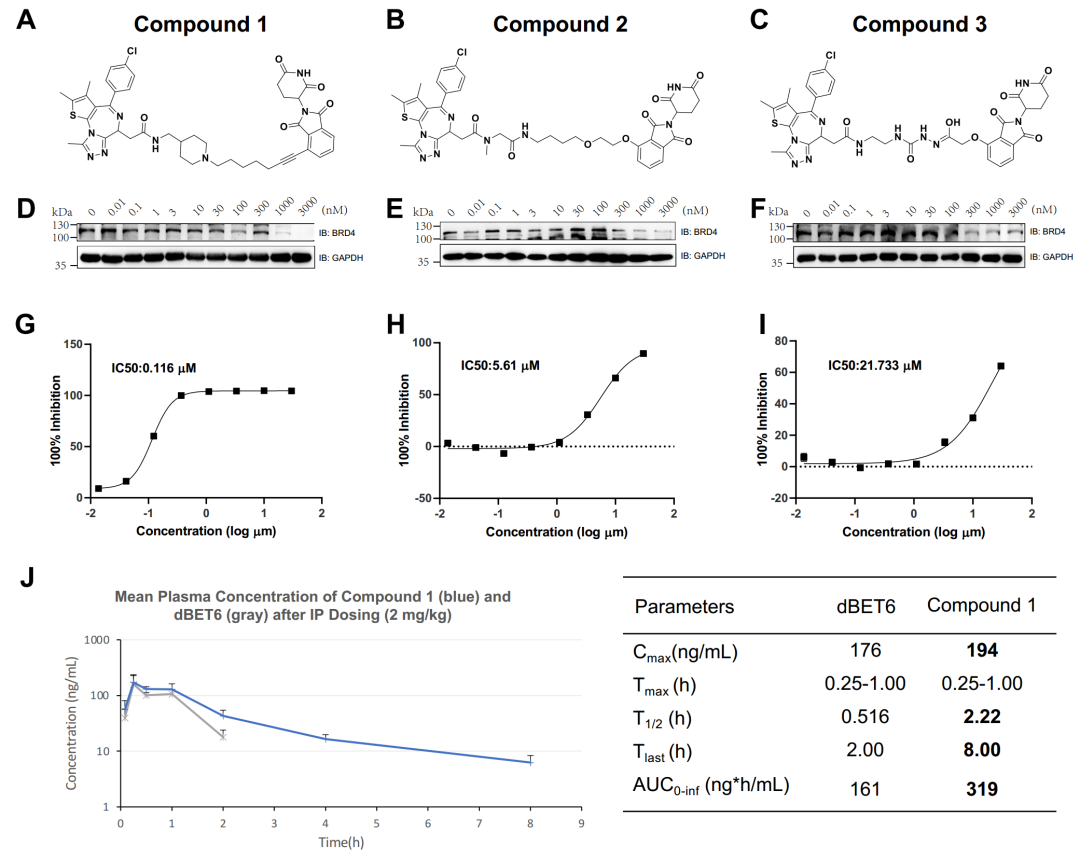
本研究报告阐述了一个完全自动化的计算框架,该框架结合了强化学习驱动的深度生成模型、机器学习和分子动力学模拟,用于合理地设计和优化PROTACs。在一项针对BRD4靶点的案例研究中,星药科技研发团队联合中山大学国家超算广州中心杨跃东教授,依托“天河二号”借助超算、AI和生物制药的交叉研究背景,充分结合基于强化学习的分子生成和基于物理的分子模拟技术,利用智能与模拟计算双驱动加速PROTAC药物研发,在短短49天内发现了具有高降解活性及高药代性能的新型先导化合物并完成湿实验验证。这进一步证明了将超算、人工智能驱动的计算策略与实验相结合是获得有效候选药物的重要手段。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
