
Science子刊:β淀粉样蛋白才是导致阿尔茨海默病的元凶!它劫持去甲肾上腺素信号来激活病理性的GSK3β/tau级联反应
来源:本站原创 2020-01-22 13:10
2020年1月22日讯/生物谷BIOON/---在一项新的临床前研究中,来自美国阿拉巴马大学伯明翰分校的研究人员发现了阿尔茨海默病的一个重要缺失部分。这使得利用一种显著降低两种小鼠模型中的阿尔茨海默病病理和症状的现有药物进行概念验证实验成为可能,从而有潜力为这种破坏性疾病提供及时治疗。相关研究结果发表在2020年1月15日的Science Translati
2020年1月22日讯/生物谷BIOON/---在一项新的临床前研究中,来自美国阿拉巴马大学伯明翰分校的研究人员发现了阿尔茨海默病的一个重要缺失部分。这使得利用一种显著降低两种小鼠模型中的阿尔茨海默病病理和症状的现有药物进行概念验证实验成为可能,从而有潜力为这种破坏性疾病提供及时治疗。相关研究结果发表在2020年1月15日的Science Translational Medicine期刊上,论文标题为“β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade”。
论文通讯作者、阿拉巴马大学伯明翰分校的Qin Wang博士说,“我们的研究针对β淀粉样蛋白(Aβ)毒性潜在机制提供了临床转化上的新见解,这可能对未来的药物设计产生重大影响。它鉴定出Aβ和G蛋白偶联受体之间的相互作用,这代表着阿尔茨海默病的一种有吸引力的疾病特异性的治疗靶标。”
有趣的是,这种新发现的发病机制可能也解释了许多阿尔茨海默病临床试验为何遭遇失败,这些临床试验靶向减少这种疾病的罪魁祸首---大脑中的Aβ蛋白堆积。
Wang说,人们普遍认为大脑中的Aβ低聚物堆积是诱导tau蛋白发生病理变化的诱因,而这种病理变化靶向并杀死阿尔茨海默病患者中的神经元。但是,连接这两者的途径尚不清楚。
Wang及其同事们发现Aβ低聚物劫持了大脑神经元中的去甲肾上腺素信号,从而错误地重新定向这种信号来激活一种称为GSK3β的激酶。这种活化的激酶接着导致tau蛋白高度磷酸化,从而使得它对神经元有毒。
去甲肾上腺素信号的这种重新定向发生于神经元表面上的一种称为α2A肾上腺素能受体(alpha-2A adrenergic receptor, α2AAR)的细胞膜受体。这种受体是一种G蛋白偶联受体。 Wang及其同事们发现,虽然一定浓度的Aβ低聚物可以激活GSK3β,但是去甲肾上腺素的存在极大地使得这种激活的灵敏度提高了两个数量级。
因此,这些研究人员推测,人大脑中的纳摩尔浓度的Aβ低聚物在阿尔茨海默病的最早阶段会诱发致病性的GSK3β/tau级联反应。这一理论提出降低阿尔茨海默病患者中Aβ低聚物水平的多项临床试验遭遇失败的原因在于它们无法将Aβ水平降低至如此低的浓度。
研究细节
α2AAR通常以以下方式起作用,即它结合去甲肾上腺素,这种结合激活了一种调动大脑和身体采取行动的信号转导过程。这些研究人员发现,Aβ低聚物与α2AAR上的一个独特位点结合,这个结合位点与去甲肾上腺素的结合位点不同。这启动了病理性劫持(pathological hijacking)。
在第二个位点上的这种结合称为变构结合(allosteric binding)。在G蛋白偶联受体中,已知变构配体通常会改变这种受体的信号转导,这是正常生理的一部分。在这些研究人员认识到变构结合后,他们进行了搜索,以发现哪种激酶可能被这种结合激活,这就是他们鉴定GSK3β的方式。
一些临床数据支持这种机制。这些研究人员发现,与非痴呆的低病理对照者相比,来自阿尔茨海默病患者死后前额叶皮层的α2AAR具有显着增加的活性。此外,对来自美国国家阿尔茨海默病协调中心(National Alzheimer's Coordinating Center)的病例进行的流行病学分析表明,服用药物可乐定(clonidine)----作为一种可降低血压的α2AAR激活剂---会恶化认知缺陷患者的认知功能。此外,可乐定对重度痴呆患者的不良反应更强。使用可乐定对具有正常认知能力的受试者没有影响。
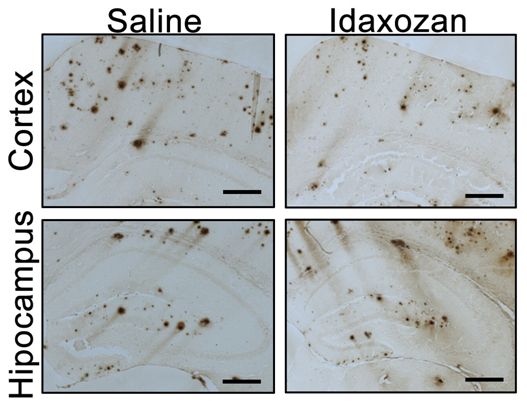
Wang及其同事们在阿尔茨海默病小鼠模型中测试了一种现有的药物:咪唑克生(idazoxan)。咪唑克生是一种α2AAR拮抗剂,已在抑郁症的临床试验中进行了研究。一种假设就是在存在Aβ病理的情况下,咪唑克生阻断α2AAR将会显示出治疗潜力。这在阿尔茨海默病小鼠模型中得到证实。
从8个月大时开始,对这些小鼠进行咪唑克生治疗8周,在开始治疗时,它们的大脑中已存在Aβ斑块,并且α2AAR显示出增强的活性。与对照者相比,这些研究人员发现:(1)咪唑克生逆转了这些小鼠大脑中的GSK3β过度活化,进一步支持了在体内,α2AAR在介导Aβ诱导的GSK3β活化中起着关键作用;(2)在接受咪唑克生治疗的阿尔茨海默病小鼠模型的大脑皮层中,Aβ负荷的程度较低,这表明阻断α2AAR减慢了Aβ病理进展;(3)咪唑克生治疗降低了炎性小胶质细胞的密度,这表明神经炎症减少;(4)咪唑克生治疗可减少tau蛋白过度磷酸化,这表明阻断α2AAR可有效缓解Aβ诱导的tau病理;(5)在两项认知功能测试中,用咪唑克生治疗的阿尔茨海默病小鼠的表现几乎与正常小鼠相当,并且明显优于未经治疗的阿尔茨海默病小鼠。
Wang说,“这些数据共同证实通过α2AAR阻断去甲肾上腺素信号是一种缓解与Aβ相关的病理和认知缺陷的有效策略。”
Wang说,“诸如咪唑克生之类的α2AAR阻断剂已被开发出来用于治疗其他的疾病,而改用这些药物可能是治疗阿尔茨海默病的潜在有效的策略。此外,我们的数据提示着Aβ与α2AAR之间的相互作用是阿尔茨海默病的一种有吸引力的疾病特异性治疗靶标,这是因为α2AAR/GSK3β/tau级联反应只能在Aβ低聚物的存在下才被激活。”
Wang说,“直接靶向Aβ/α2AAR的变构界面不会干扰正常的α2AAR功能,因此不太可能导致与治疗阿尔茨海默病所必需的长时间给药相关的并发症。”(生物谷 Bioon.com)
参考资料:
1.Fang Zhang et al. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Science Translational Medicine, 2020, doi:10.1126/scitranslmed.aay6931.
2.Pathogenic Alzheimer's disease cascade is activated by faulty norepinephrine signaling
https://medicalxpress.com/news/2020-01-pathogenic-alzheimer-disease-cascade-faulty.html
图片来自Science Translational Medicine, 2020, doi:10.1126/scitranslmed.aay6931。
论文通讯作者、阿拉巴马大学伯明翰分校的Qin Wang博士说,“我们的研究针对β淀粉样蛋白(Aβ)毒性潜在机制提供了临床转化上的新见解,这可能对未来的药物设计产生重大影响。它鉴定出Aβ和G蛋白偶联受体之间的相互作用,这代表着阿尔茨海默病的一种有吸引力的疾病特异性的治疗靶标。”
有趣的是,这种新发现的发病机制可能也解释了许多阿尔茨海默病临床试验为何遭遇失败,这些临床试验靶向减少这种疾病的罪魁祸首---大脑中的Aβ蛋白堆积。
Wang说,人们普遍认为大脑中的Aβ低聚物堆积是诱导tau蛋白发生病理变化的诱因,而这种病理变化靶向并杀死阿尔茨海默病患者中的神经元。但是,连接这两者的途径尚不清楚。
Wang及其同事们发现Aβ低聚物劫持了大脑神经元中的去甲肾上腺素信号,从而错误地重新定向这种信号来激活一种称为GSK3β的激酶。这种活化的激酶接着导致tau蛋白高度磷酸化,从而使得它对神经元有毒。
去甲肾上腺素信号的这种重新定向发生于神经元表面上的一种称为α2A肾上腺素能受体(alpha-2A adrenergic receptor, α2AAR)的细胞膜受体。这种受体是一种G蛋白偶联受体。 Wang及其同事们发现,虽然一定浓度的Aβ低聚物可以激活GSK3β,但是去甲肾上腺素的存在极大地使得这种激活的灵敏度提高了两个数量级。
因此,这些研究人员推测,人大脑中的纳摩尔浓度的Aβ低聚物在阿尔茨海默病的最早阶段会诱发致病性的GSK3β/tau级联反应。这一理论提出降低阿尔茨海默病患者中Aβ低聚物水平的多项临床试验遭遇失败的原因在于它们无法将Aβ水平降低至如此低的浓度。
研究细节
α2AAR通常以以下方式起作用,即它结合去甲肾上腺素,这种结合激活了一种调动大脑和身体采取行动的信号转导过程。这些研究人员发现,Aβ低聚物与α2AAR上的一个独特位点结合,这个结合位点与去甲肾上腺素的结合位点不同。这启动了病理性劫持(pathological hijacking)。
在第二个位点上的这种结合称为变构结合(allosteric binding)。在G蛋白偶联受体中,已知变构配体通常会改变这种受体的信号转导,这是正常生理的一部分。在这些研究人员认识到变构结合后,他们进行了搜索,以发现哪种激酶可能被这种结合激活,这就是他们鉴定GSK3β的方式。
一些临床数据支持这种机制。这些研究人员发现,与非痴呆的低病理对照者相比,来自阿尔茨海默病患者死后前额叶皮层的α2AAR具有显着增加的活性。此外,对来自美国国家阿尔茨海默病协调中心(National Alzheimer's Coordinating Center)的病例进行的流行病学分析表明,服用药物可乐定(clonidine)----作为一种可降低血压的α2AAR激活剂---会恶化认知缺陷患者的认知功能。此外,可乐定对重度痴呆患者的不良反应更强。使用可乐定对具有正常认知能力的受试者没有影响。
Wang及其同事们在阿尔茨海默病小鼠模型中测试了一种现有的药物:咪唑克生(idazoxan)。咪唑克生是一种α2AAR拮抗剂,已在抑郁症的临床试验中进行了研究。一种假设就是在存在Aβ病理的情况下,咪唑克生阻断α2AAR将会显示出治疗潜力。这在阿尔茨海默病小鼠模型中得到证实。
从8个月大时开始,对这些小鼠进行咪唑克生治疗8周,在开始治疗时,它们的大脑中已存在Aβ斑块,并且α2AAR显示出增强的活性。与对照者相比,这些研究人员发现:(1)咪唑克生逆转了这些小鼠大脑中的GSK3β过度活化,进一步支持了在体内,α2AAR在介导Aβ诱导的GSK3β活化中起着关键作用;(2)在接受咪唑克生治疗的阿尔茨海默病小鼠模型的大脑皮层中,Aβ负荷的程度较低,这表明阻断α2AAR减慢了Aβ病理进展;(3)咪唑克生治疗降低了炎性小胶质细胞的密度,这表明神经炎症减少;(4)咪唑克生治疗可减少tau蛋白过度磷酸化,这表明阻断α2AAR可有效缓解Aβ诱导的tau病理;(5)在两项认知功能测试中,用咪唑克生治疗的阿尔茨海默病小鼠的表现几乎与正常小鼠相当,并且明显优于未经治疗的阿尔茨海默病小鼠。
Wang说,“这些数据共同证实通过α2AAR阻断去甲肾上腺素信号是一种缓解与Aβ相关的病理和认知缺陷的有效策略。”
Wang说,“诸如咪唑克生之类的α2AAR阻断剂已被开发出来用于治疗其他的疾病,而改用这些药物可能是治疗阿尔茨海默病的潜在有效的策略。此外,我们的数据提示着Aβ与α2AAR之间的相互作用是阿尔茨海默病的一种有吸引力的疾病特异性治疗靶标,这是因为α2AAR/GSK3β/tau级联反应只能在Aβ低聚物的存在下才被激活。”
Wang说,“直接靶向Aβ/α2AAR的变构界面不会干扰正常的α2AAR功能,因此不太可能导致与治疗阿尔茨海默病所必需的长时间给药相关的并发症。”(生物谷 Bioon.com)
参考资料:
1.Fang Zhang et al. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Science Translational Medicine, 2020, doi:10.1126/scitranslmed.aay6931.
2.Pathogenic Alzheimer's disease cascade is activated by faulty norepinephrine signaling
https://medicalxpress.com/news/2020-01-pathogenic-alzheimer-disease-cascade-faulty.html
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
87%用户都在用生物谷APP 随时阅读、评论、分享交流 请扫描二维码下载->
