
艾伯维修美乐治疗中重度活动性溃疡性结肠炎儿童患者获FDA批准
来源:新浪医药新闻 2021-02-26 13:04
日前,艾伯维宣布,美国FDA批准Humira(阿达木单抗,商品名修美乐)用于治疗5岁及以上儿童中度至重度活动性溃疡性结肠炎(UC)。艾伯维介绍,此次批准是基于关键的3期临床研究ENVISION I的结果。该研究表明,Humira在第8周达到了部分Mayo评分(PMS)临床缓解的共同主要终点,在第8周有反应的患者中,在一年(52周)达到
日前,艾伯维宣布,美国FDA批准Humira(阿达木单抗,商品名修美乐)用于治疗5岁及以上儿童中度至重度活动性溃疡性结肠炎(UC)。
艾伯维介绍,此次批准是基于关键的3期临床研究ENVISION I的结果。该研究表明,Humira在第8周达到了部分Mayo评分(PMS)临床缓解的共同主要终点,在第8周有反应的患者中,在一年(52周)达到了完全Mayo评分(FMS)临床缓解的共同主要终点。临床缓解被定义为PMS或FMS小于或等于2,且个体得分不大于1。
ENVISION I是一项随机、双盲、多中心3期研究,旨在评估Humira在中重度UC患儿(年龄4-17岁)中的疗效、安全性和药代动力学(定义为FMS为6-12,内窥镜检查得分为2-3分,经中央阅读内窥镜检查证实),皮下注射。
到第8周,两个剂量组的患者在第0周服用Humira 2.4 mg/kg(最大160 mg),第2周服用1.2 mg/kg(最大80 mg),第4周和第6周服用0.6 mg/kg(最大40 mg)。高剂量组在第1周还接受了2.4 mg/kg(最大160 mg)的额外剂量。在第8周和第52周之间,患者每隔一周或每周接受双盲安慰剂,Humira 0.6 mg/kg(最多40 mg)。该研究的共同主要终点是在第8周时每经前综合症的临床缓解率(定义为经前综合症≤2分且无个体分项评分>1),以及在第52周时每经前综合症达到临床缓解率(定义为Mayo评分≤2分且无个体分项评分>1)患者Mayo评分的临床缓解率(定义为Mayo评分≤2分且无个体分项评分>1分)。
研究结果显示,在8周诱导期结束时,服用高剂量Humira的患者中,有60%的患者(28/47)在每次经前综合症时获得临床缓解,而服用低剂量Humira的患者中,有43%的患者(13/30)获得临床缓解。在第52周,在第8周的PMS应答者中,接受高剂量Humira治疗的患者中,45%的患者在每次FMS中病情缓解,接受低剂量Humira治疗的患者中,有29%的患者病情缓解,随机接受安慰剂治疗的患者中,有33%的患者病情缓解。由于样本量小,安慰剂数据的可解释性受到限制。
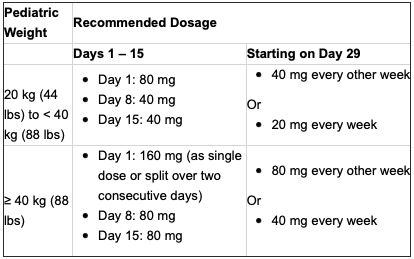
Humira的批准剂量将根据儿童的体重确定,如下:
对于年满18岁的患者,以及对其Humira治疗方案控制良好的患者,建议继续使用推荐的儿科剂量。
在ENVISION I研究中,没有观察到Humira的新安全信号。在该研究中的任何Humira暴露期间,22.6%的患者经历了严重的不良事件。诱导期和维持期最常报告的(大于或等于5%)因治疗引起的不良事件是头痛和溃疡性结肠炎恶化。本研究未观察到死亡、恶性肿瘤、活动性肺结核或脱髓鞘疾病。
2018年,Humira是全球最畅销的处方药,这一趋势持续了几年。该药物仅在2018年就为艾伯维带来了约200亿美元的收入。尽管在2023年之前,生物仿制药还无法进入美国市场,Humira获得了专利保护,但在2018年末,欧洲出现的生物仿制药竞争对手,包括诺华、安进、迈兰和三星Bioepis都开发了Humira的生物仿制药,全球销量直线下跌。
2021年12月,艾伯维宣布了另一种中重度UC药物Rinvoq(upadacitinib)的3期诱导研究U-ACHIEVE的阳性结果。该试验在8周时达到了成年患者临床病情缓解的主要终点。另一项3期诱导研究U-ACCOMPLISH也证实,Rinvoq改善了UC患者的临床、内镜和组织学结果。(生物谷Bioon.com)
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
