
Science:一种发现人类基因组中结构变异的新方法消除了现有人类基因分型中存在的偏差
来源:本站原创 2021-12-20 20:30
科学家们早就认识到,单一参考基因组不能代表人类的多样性,而且使用它给这些研究带来普遍的偏见。如今,在一项新的研究中,来自美国加州大学圣克鲁兹分校和田纳西大学等研究机构的研究人员终于有了一个实用的替代方案。
2021年12月19日讯/生物谷BIOON/---自从20多年前首次对人类基因组进行测序以来,对人类基因组的研究几乎完全依赖于单一的参考基因组,并与其他基因组进行比较,以确定遗传变异。科学家们早就认识到,单一参考基因组不能代表人类的多样性,而且使用它给这些研究带来普遍的偏差。如今,在一项新的研究中,来自美国加州大学圣克鲁兹分校和田纳西大学等研究机构的研究人员终于有了一个实用的替代方案。相关研究结果发表在2021年12月17日的Science期刊上,论文标题为“Pangenomics enables genotyping of known structural variants in 5202 diverse genomes”。
在这篇论文中,这些作者介绍了一种称为Giraffe的新工具,它可以有效地将新的基因组序列映射到代表许多不同人类基因组序列的“泛基因组(pangenome)”。他们表明,这种方法可以更全面地描述遗传变异的特征,并可以改善众多研究人员和临床医生所使用的基因组分析。
论文通讯作者、加州大学圣克鲁兹分校生物分子工程副教授Benedict Paten说,“我们多年来一直朝着这个方向努力,如今我们第一次有了一些实用的东西,它可以快速工作,而且比单一参考基因组的效果更好。对于生物医学的未来来说,基因组学平等地帮助每个人是很重要的,所以我们需要考虑到人类群体的多样性并且不存在偏差的工具。”
所有的人都有相同的基因,但在这些基因的确切序列以及在蛋白质编码基因之外的大部分基因组序列中存在许多变化。基因组中单个碱基的差异称为单核苷酸变异(SNV),而短序列的插入或缺失被统称为indel。
最复杂的变异是涉及大片段序列(50个或更多碱基)重新排列的结构变异。使用单一参考基因组尤其难以发现这些变异,但它们可以产生重大影响,并且已知在一些疾病中发挥重要作用。普通人有数以百万计的SNV和indel以及数以万计的更大的结构变异,而且总体上,结构变异实际上比其他类型的变异涉及更多的碱基序列。
Paten说,“基因组学研究一直聚焦于SNV和indel,因为结构变异一直被隐藏起来。泛基因组学正在使结构变异变得可见,因此我们可以像研究SNV和indel一样研究它们。有很多结构变异,它们可以产生很大的影响,所以这对疾病的遗传研究的未来至关重要。”
作为参考的泛基因组可以从多个基因组序列中创建,使用数学图形结构来表示不同序列之间的关系。在这篇新论文中,这些作者利用公开的数据建立了两个人类基因组参考图。这些可被用来评估新工具Giraffe,其中Giraffe是一组用于将新的序列数据映射到参考泛基因组的算法。
论文共同第一作者、加州大学圣克鲁兹分校基因组学研究所研究员Jouni Sirén开创了Giraffe的许多关键算法创新。Giraffe能够准确地将新的序列数据映射到嵌入参考泛基因组中的数千个基因组上,就像现有工具映射到单个参考基因组一样迅速。该研究还显示,使用Giraffe减少了映射偏差,即错误地映射与参考基因组不同的序列的倾向。
论文共同第一作者、加州大学圣克鲁兹分校基因组学研究所博士后研究员Jean Monlong说,“它不仅分析结果更好,而且与目前使用线性参考基因组的方法一样快。”
廉价的短读测序(short-read sequencing)是现代基因组学的主流,产生的序列片段必须映射到参考基因组上才能理解。映射显示每个片段属于人类23条染色体中的哪一条,并确定个人基因组中每个位置存在的变异,这一过程被称为基因分型。
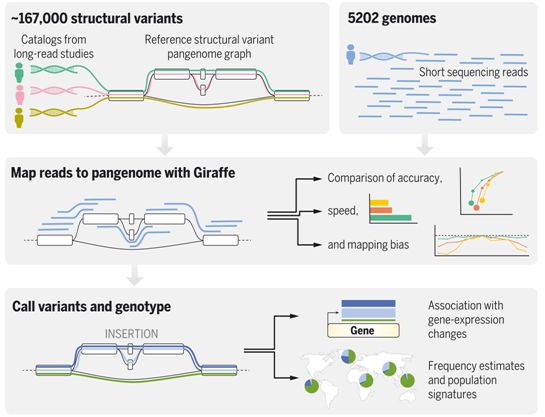
实验概述:来自基于长读测序的和大规模测序研究的变体调用被用来构建泛基因组参考图(顶部)。Giraffe(和竞争的映射程序)将读取序列映射到泛基因组参考图或线性参考基因组上,并对映射的准确性、等位基因覆盖平衡和速度进行评估(中间)。然后,映射的读取序列被用于变异调用,并对变异调用的准确性进行了评估(底部)。结构变异调用与表达数据一起进行分析,以确定eQTL和群体频率估计。图片来自Science, 2021, doi:10.1126/science.abg8871。
这些作者发现,Google Health的深度学习变体调用器DeepVariant,使用Giraffe针对泛基因组的比对,比使用针对单一参考基因组的比对能更准确地识别SNV和 indel。
Monlong说,他对使用泛基因组学来研究结构变异感到最兴奋。他说,“最近使用长读测序(long-read sequencing)发现了很多结构变异。有了泛基因组,我们可以在短读测序的大型数据集中寻找这些结构变异。这很令人兴奋,因为这将使我们能够在许多人中研究这些新的结构变异,并提出有关其功能影响、与疾病的关联或在进化中的作用的问题。”
这些作者用Giraffe对来自5202人的不同群体的读取序列进行了映射,并确定了16.7万个最近发现的结构变异的基因型。这使他们能够估计这些结构变异的不同版本在整个人类群体和不同亚群中的频率。他们发现,一些结构变异的频率在不同的亚群之间有很大的差异,如果仅在欧洲血统的人群中分析,它们可能会被误解,因为该人群中的特定结构变异频率很低。
单一的参考基因组必须选择任何变异的一个版本来代表,而使其他的版本没有得到代表。通过使更具广泛代表性的参考泛基因组成为现实,Giraffe可以使基因组学更具包容性。
Paten和加州大学圣克鲁兹分校基因组学研究所的其他人参与了一项由美国国家人类基因组研究中心资助的重大努力,以便建立全面的人类参考泛基因组,他们预计将在明年发布它,作为科学界的一个资源。(生物谷 Bioon.com)
参考资料:
Jouni Sirén et al. Pangenomics enables genotyping known structural variants in 5,202 diverse genomes. Science, 2021, doi:10.1126/science.abg8871.
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
