
2023年,FDA发出36份CRL!临床数据缺乏、CMC缺陷占大头
来源:医药魔方 2023-12-17 10:12
通过对2023年FDA发出的CRL可以发现,临床数据缺陷和CMC问题是影响到药物获批的最主要原因。
在美国的制药监管实践中,完整回应函(CRL),或更罕见的314.110信件,是美国对新药申请(NDA)、修订的新药申请或生物制品许可申请(BLA)作出回应的监管行动,表明该申请将不会以目前的形式获得批准。2008年,FDA修改新药和仿制药审批规定,以完整回应函(complete response letter,CRL)取代原本的可批准/不可批准信函。
CRL实则是一封拒信,意为FDA已经完成审查,但现有的申请不满足批准要求。但是,这也不代表着FDA完全拒绝该款药物上市,FDA通常会在CRL中详细说明药物可能存有的缺陷和风险,并提出建议和改进方案。如果申请人能在规定时间内完成更改,这款药物仍能够获批上市。
为了提高监管过程的整体透明度,FDA曾就是否公开这些CRL展开过讨论,不过,由于涉及到药品相关的机密信息,遭到了制药行业人员的反对。因此,FDA通常只将CRL单独发送给企业。
根据企业公开披露的信息整理,2023年以来FDA累计发出了36份完整回复函(CRL),其中有近一半(47%)来自创新药NDA或者BLA申请;有8份来自仿制药/生物类似药上市申报,也有6款新药在申报新适应症的过程中收到了FDA发出的CRL;此外,有5款改良型新药在申报新剂型、新规格的过程中收到了CRL。
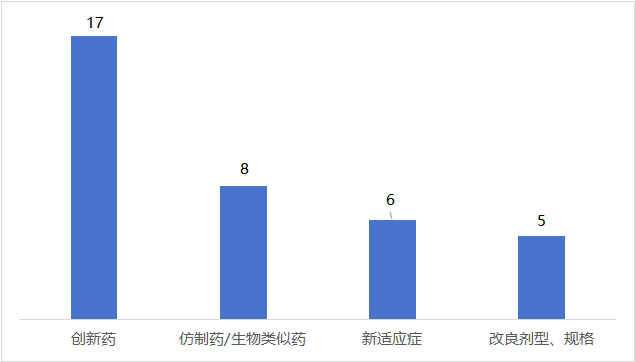
FDA发布CRL所涉及的药物类型
36份CRL中,既涉及小分子药物,也涉及到多肽、融合蛋白,单克隆抗体等大分子生物药;这些收到CRL的企业中,既包括礼来、阿斯利康、诺和诺德、再生元等跨国制药企业,也涉及一些小型Biotech,不过Biotech通常占多数。
充分的临床数据是支持新药批准的基石
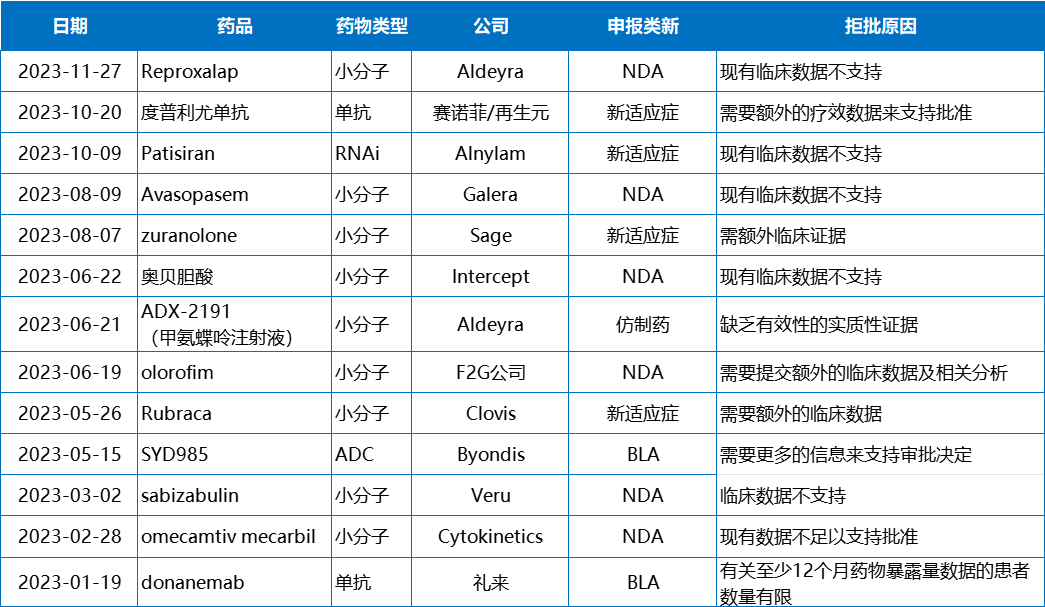
CRL相当于FDA“手握”的生杀大权,有时候决定着一款药物、甚至一个企业的生死。临床有效性和安全性数据通常也与这款药物的“命运”直接挂钩。2023年,CRL中提及“现有临床数据缺乏,不足以支持批准”的药物数量占到了所有CRL数量的39%(14/36)。
年初,礼来曾基于II期研究数据向FDA递交了阿尔兹海默病(AD)新药donanemab的加速上市申请,不过年初收到了FDA的CRL,FDA要求礼来提供至少100例接受过至少12个月donanemab持续治疗的患者数据。好在donanemab的III期研究于今年7月获得了积极结果,礼来也已于第二季度完成向FDA递交上市申请,预计年底收到反馈。
相较于礼来,Cytokinetics则没有那么幸运了。在经历了19年漫长研发之后,Cytokinetics公司的first in class心衰药omecamtiv mecarbil仍换得了FDA冰冷的CRL。紧接着,这款药物在中国的NDA申请同样吃了NMPA的“闭门羹”。祸不单行,4月初,由于候选药物reldesemtiv未通过二次期中分析,Cytokinetics终止了该药物治疗肌萎缩侧索硬化(ALS)的III期COURAGE-ALS研究。两款后期管线接连失败,Cytokinetics受到重创,今年以来股价持续走跌。
奥贝胆酸(Ocaliva)曾被认为是40多年NASH困局将要迎来的第一缕阳光。然而,在经历了重新递交NDA申请、专家咨询委员会讨论后,6月22日FDA下发的CRL彻底浇灭了Intercept的梦。FDA表示目前的数据不支持奥贝胆酸获得批准,还需要额外的III期研究数据。这也导致Intercept终止了所有NASH相关投资,并最终以8亿美元的现金将公司出售。
除此之外,IL-4/IL-13单抗度普利尤单抗、RNAi疗法Patisiran、PARP抑制剂Rubraca(鲁卡帕尼)曾因新适应症申报过程中,需要更多临床数据而收到FDA下发的CRL。
2023年因临床数据不足而收到CRL的药物
涉及到药物安全性和有效性的临床数据是证明一款药物是否对患者有获益的准绳。也是FDA把关的“重阵”。对于那些有临床治疗变革性的药物,或是极具争议的药物,FDA通常还会召开专家咨询委员会以听取他们的意见。虽然FDA有独立于他们的意见做出审评结论的权利,不过从过往批准情况来看,专家委员会的意见具有十分重要的参考价值。
面对FDA严格的审查流程和标准,仍有一些“幸运儿”顺利获FDA批准上市,包括一款渐冻症药物Relyvrio(苯丁酸钠和牛磺酸二醇口服固定剂量配方,AMX0035)。专家委员会曾以6:4的投票,表达了对AMX0035现有数据说服力的怀疑。不过,在新一轮的委员会会议中,专家们仍以7:2的投票结果赞成AMX0035。这也是主要考虑到针对ALS疾病,现有疗法十分有限。
一位行业人士也曾表示:“相较于其他问题,FDA更看重一款新药的安全性和有效性问题。这是因为FDA肩负着双重使命,他们既需要考虑临床未满足的需求,为美国人民找到可供使用药物的责任,也有对新药质量进行把关的责任,因此FDA会从中找到risk和benefit之间的平衡。而对于那些尚无药物可治疗的疾病来说,对临床数据的审核更为关键。”
CMC问题同样不容小觑
2023年,因药物化学、生产与控制(Chemistry Manufacturing & Control,CMC)问题而收到CRL的药物占比高达50%(18/36)。从药物类型上来看,相较于小分子药物,生物药因CMC问题收到CRL的概率更高(4款 vs. 14款),成为接收CRL的“重灾区”。这也主要和生物药原料制备更复杂、质量控制难度更高有关。
因CMC问题收到CRL的药物
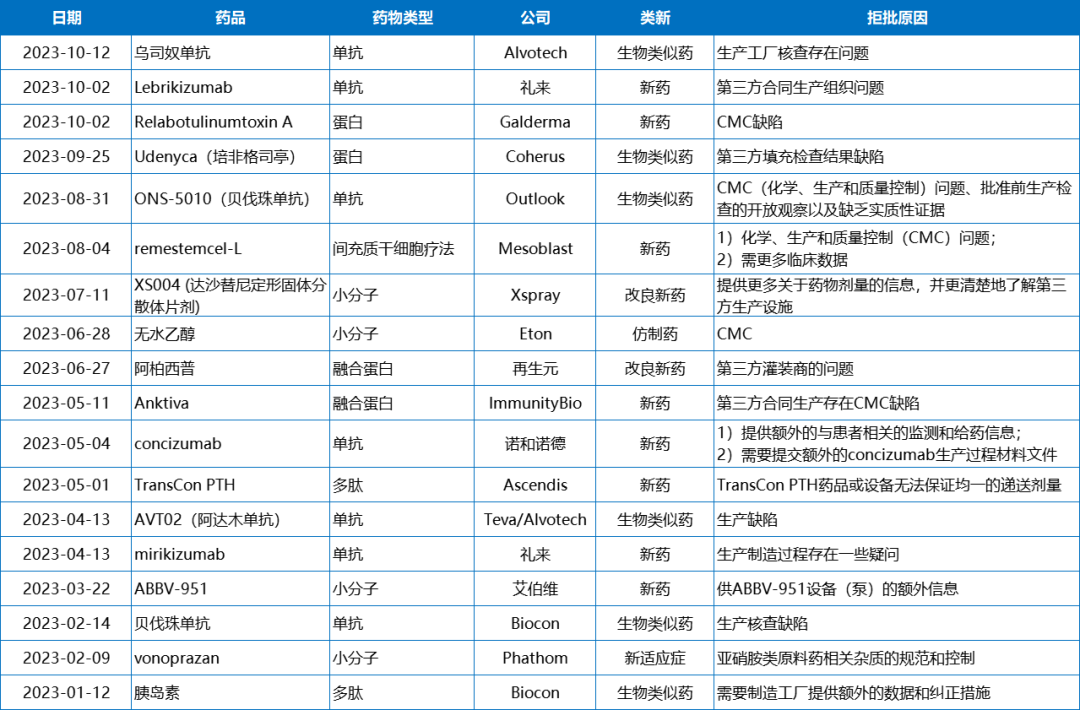
礼来的特应性皮炎新药Lebrikizumab因为在检查中发现了第三方合同生产组织的问题,IL-23p19单抗mirikizumab也因生产制造过程中存在的问题收到CRL。ImmunityBio公司的膀胱癌治疗药物Anktiva同样因第三方合同制造商存在缺陷而收到CRL,这也导致该公司股价在盘前交易中下跌了近60%。此后,该公司解雇了50名员工以“简化运营”,专注于“当前的业务战略并降低成本”。
进一步研究我们也发现因第三方合同制造商导致的CMC问题十分普遍。除了礼来和ImmunityBio外,再生元所递交的阿柏西普8mg制剂的上市申请、Coherus公司的培非格司亭生物类似药均因第三方生产问题收到CRL。这也提醒我们即使产品生产进行委外开发,对第三方的监管和核查需要引起高度重视。
此外,即使是有原研产品作为参考,生物类似药因CMC问题而遭到FDA拒绝也十分常见。具体而言,Outlook公司开发的眼科制剂贝伐珠单抗生物类似药曾因CMC问题而遭到FDA拒绝,这也导致Outlook股价大跌80%;10月,Alvotech开发的乌司奴单抗生物类似药AVT04也因为生产工厂核查存在问题遭到FDA冷落,这也是今年该公司第二次遭到FDA的拒绝,今年初,其开发的阿达木单抗生物类似药AVT02因生产缺陷收到了FDA的CRL。
还有少部分因安全性问题、产品检测不合格以及需补充非临床试验而收到CRL的药物。9月份,长效C5抑制剂Ultomiris(ravulizumab)的sBLA收到了FDA的CRL,FDA要求阿斯利康修改Ultomiris的风险评估和减轻策下发给略(REMS)。REMS是一种关于药物的风险管理计划,以帮助确保药物的获益大于其风险。在发送给Citius公司的CRL中,FDA要求该公司加强对工程化IL-2白喉毒素融合蛋白denileukin diftitox 的产品测试,并在上市申请审查期间与FDA达成额外的控制协议。
总结
通过对2023年FDA发出的CRL可以发现,临床数据缺陷和CMC问题是影响到药物获批的最主要原因。不过这两类问题也都不会导致产品的最终失败,FDA发出整改建议后,企业也会根据产品和公司的实际情况做出继续开发或者终止开发的决定。
另一方面,我们也能够感知到相较于大企业来说,由于产品群丰富,单个的产品拒批不会影响到公司的整体发展,不过对于小型制药企业来说,是牵一发而动全身,一款产品的申报失败也可能直接导致公司的运营陷入危机,伴随而来的裁员,甚至倒闭、卖身也是常有的事。这也告诉我们,每一款产品的开发需要每一步都走得稳妥,不可抱有侥幸心理。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
