
全基因组分析如何助力人类疾病研究?
来源:生物谷原创 2022-05-06 15:44
本文中,小编整理了多篇重要研究成果,共同解读科学家们如何进行全基因组分析来助力人类疾病的研究,分享给大家!
本文中,小编整理了多篇重要研究成果,共同解读科学家们如何进行全基因组分析来助力人类疾病的研究,分享给大家!
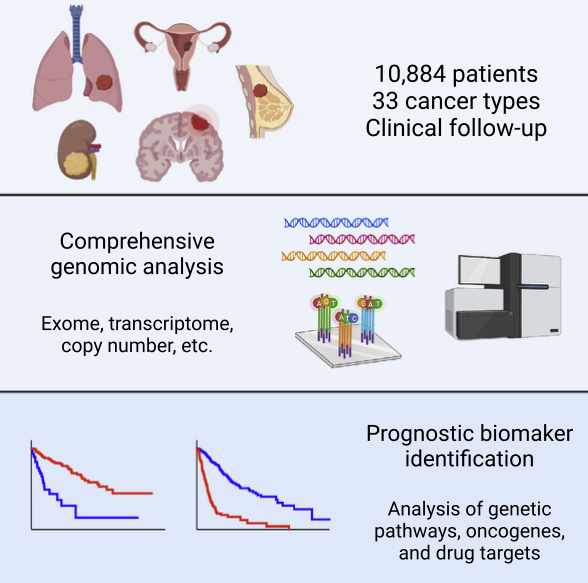
全基因组鉴定和分析人类癌症的预后特征
图片来源: https://doi.org/10.1016/j.celrep.2022.110569
【1】Cell Reports:全基因组鉴定分析人类癌症的预后生物标志物
doi:10.1016/j.celrep.2022.110569
准确区分侵袭性癌症和惰性癌症的能力是预测患者风险的基础,并可以指导关键的治疗决策。对于良性癌症,谨慎等待和/或手术切除可能是合适的,而浸润性癌症可能需要使用细胞毒疗法进行多模式治疗,这种疗法本身就会导致相当大的发病率。癌症治疗不足和癌症过度治疗都已被确定为患者死亡的重要来源,突显出迫切需要提高我们准确识别患有最具侵袭性恶性肿瘤的患者的能力。目前的风险预测在很大程度上依赖于对疾病状态的组织病理学和放射学评估。淋巴结转移和细胞去分化等特征的存在被认为是患者预后的强有力的预测指标,并被用来确定癌症的分期和分级。然而,这些病理标记物需要主观判断,并且观察者之间的符合率较低。此外,即使是完美的肿瘤分期也不能明确地预测患者随后的临床病程。
近日,耶鲁大学医学院的研究者们在Cell Reports杂志上发表了题为“Genome-wide identification and analysis of prognostic features in human cancers”的文章,该研究分析为预后生物标记物分析建立了丰富的资源,并阐明了患者生存数据在临床前癌症研究和治疗开发中的应用。癌症的临床决策依赖于对患者风险的准确评估。为了提高我们识别最具侵袭性的恶性肿瘤的能力,研究者使用来自10,884名患者的基因表达、拷贝数、甲基化和突变数据构建了全基因组生存模型。
研究者确定了100,000多个重要的预后生物标记物,并证明这些基因组特征可以在临床上不明确的情况下预测患者的预后。虽然不利的生物标记物通常被认为代表癌症驱动基因和有希望的治疗靶点,但研究者表明,与较短生存时间相关的癌症特征不会对癌基因或成功的药物靶点产生丰富作用。相反,最强的不良生物标志物代表了广泛表达的细胞周期和基因,相应地,几乎所有针对这些特征的治疗都在临床试验中失败了。
【2】Science:在迄今最大规模的全基因组测序数据中鉴定替换突变特征
doi:10.1126/science.abl9283
突变特征(mutational signature,也译为突变信号、突变标记或突变签名)---在肿瘤发生过程中起作用的DNA损伤和修复过程的印记---使人们了解每名患者所患癌症的环境原因和内源性原因。癌症基因组测序研究允许对突变特征进行探索。在一项新的研究中,来自英国剑桥大学的研究人员调查了多种肿瘤类型的大量经过全基因组测序的癌症,比以前的研究多得多,以便全面加强他们对突变特征的理解。相关研究结果发表在2022年4月22的Science期刊上,论文标题为“Substitution mutational signatures in whole-genome–sequenced cancers in the UK population”。
这些作者对通过英国国家卫生服务系统(NHS)为十万人基因组计划(100,000 Genomes Project)前瞻性地收集的12222例经过全基因组测序的癌症进行突变特征分析。他们在每个器官中独立鉴定了单碱基替换(SBS)和双碱基替换(DBS)突变特征。利用这个特别庞大的队列,他们开发了一种方法来加强对常见突变过程和罕见的、低频率的突变过程的区分。
这些作者通过对两个公开的队列的数据---国际癌症基因组联盟(International Cancer Genome Consortium, ICGC)的3001例原发性癌症和哈特维希医学基金会(Hartwig Medical Foundation)的3417例转移性癌症---进行独立分析来验证他们的发现。他们通过比较和对比独立得出的组织特异性突变特征,并进行聚类分析,将来自不同组织的可能由类似过程引起的突变特征联合起来,从而产生了一组参考突变特征(reference signature)。
【3】Nat Commun:全基因组测序揭示结核分枝杆菌的抗生素“预抗性”特征!
doi:10.1038/s41467-021-27616-7
细菌全基因组测序的最新进展让研究人员成功绘制出了结核分枝杆菌抗生素耐药基因组特征的完整目录;近日,一篇发表在国际杂志Nature Communications上题为“Genomic signatures of pre-resistance in Mycobacterium tuberculosis”的研究报告中,来自帝国理工学院等机构的科学家们通过研究首次发现了细菌中“预抗性”(pre-resistance)存在的迹象,相关研究结果或能帮助临床医生未来选择针对细菌性感染的最佳疗法。
文章中,研究人员对3000多份结核病样本进行全基因组测序,并在近20年里来追踪患者的结核病感染情况;结核分枝杆菌(MTB)是一种影响肺部功能的细菌性感染性疾病,2020年在传染病引发的死亡病例中其所引发的死亡仅次于COVID-19;如果使用正确的抗生素进行治疗的话,结核病患者就能被治愈,但治疗的时间很长,而且很多患者会面临无法获得足够医疗保健的风险,如果患者无法完成整个治疗过程,或者没有药物以及药物质量较差的话,就会出现耐药性结核病的发生。
多重耐药性的结核病是一种巨大且不可持续的人类疾病负担,如今研究人员在少数国家已经发现了完全耐药的菌株,由于卫生系统正在努力应对当前新冠疫情,全球结核病治疗的进展已经大大放缓了。为了能够更好地理解并最终开发治疗结核病的新型疗法,这篇研究报告中,研究人员首次揭示了如何在耐药性突变发生之前预防结核病患者所出现的耐药性,研究人员将这一概念称之为“预抗性”,即当诸如病毒或细菌等致病微生物在未来产生耐药性的内在风险更大时。

人类癌症中体细胞突变模式的全基因组概要。
图片来源:Science, 2022, doi:10.1126/science.abg5601。
【4】Science:对癌症中体细胞非编码突变模式的全基因组分析
doi:10.1126/science.abg5601
肿瘤产生的一个关键标志是癌细胞在其基因组中获得了正常组织中不存在的体细胞突变。一些突变是驱动突变(driver mutation),有助于肿瘤细胞生长,但其他许多突变是乘客突变(passenger mutation),对肿瘤生物学没有明显影响。在过去的十年里,通过分析成千上万对肿瘤-正常组织的测序数据,驱动突变在蛋白编码基因组区域(即编码蛋白的基因组区域)中被全面表征。对蛋白编码基因组区域的这种表征产生了对肿瘤生物学的大量见解,包括许多受基因组启发的药物靶标。然而,体细胞突变在剩下98%的癌症基因组---非编码基因组(noncoding genome)---中的作用仍未被完全理解。
许多统计学方法通过比较每个基因中对蛋白编码序列有影响和无影响的突变数量,将驱动突变检测为复发性突变事件。因此,这些方法不适用于蛋白编码区域以外的地方,在那里,体细胞突变的作用仍然不甚明了。非编码基因组包括多种不同的序列元件,包括基因表达的调节区域,这些调节区域的位置和活性在不同的肿瘤类型中有所不同。在一项新的研究中,为了扩大对蛋白编码区域以外的突变的理解,来自布罗德研究所、丹娜-法伯癌症研究所、哈佛医学院、布莱根妇女医院和乌特勒支大学医学中心的研究人员设计并实施了一种全基因组的滑动窗口方法来检测突变事件,而不考虑其在调节元件中的位置或对蛋白编码序列的影响。相关研究结果发表在2022年4月8日的Science期刊上,论文标题为“Genome-wide analysis of somatic noncoding mutation patterns in cancer”。
这些作者开发了三种方法的组合,以检测含有6120万个体细胞突变的19种癌症类型的3949名患者的全基因组中的复发性突变事件。这种方法根据突变事件在基因组中的位置,自动将其分为不同的类别。在蛋白编码区域,他们发现每种癌症类型平均有7.5个事件,并获得了公认的驱动突变。在非编码基因组中,每种癌症类型有3.7个事件发生在特定组织类型中专门表达的基因附近(肝脏中的ALB基因,前列腺中的KLK3基因,肺部中的SFTPB基因,肾脏中的SLC5A12,甲状腺组织中的TG基因,以及其他)。这些组织特异性事件不太可能是典型的驱动突变,因为它们源于只在这些基因周围活跃的诱变过程,而不是反映了肿瘤细胞起源的表达程序的可能印记。
【5】PNAS:全基因组筛查肠道微生物在调节神经退行性疾病中的研究
doi:10.1073/pnas.2106504118
越来越多的证据表明,肠道微生物在调节神经退行性疾病(如帕金森病)的进展方面发挥着关键作用。这种微生物与宿主相互作用的分子机制还不清楚。在本研究中,作者通过用大肠杆菌敲除突变体喂养表达人α-syn的秀丽隐杆线虫,进行了全基因组筛选,以确定促进宿主神经变性的细菌基因。
神经退行性疾病的特征是蛋白质错误折叠和聚集,导致β-片结构丰富的淀粉样原纤维的形成。这种蛋白质聚集引发蛋白质毒性,最终导致神经元死亡。DA神经元的缺失导致纹状体多巴胺信号的减少,从而导致PD患者运动功能受损。α-syn是一个小的(140个氨基酸)蛋白质,由一个n端结构域、一个非a -β组成的结构域(是纤维化核心)和一个羧基末端区域组成。n端结构域的错义突变,如A30P、G46K和A53T,通过产生比野生型更容易错误折叠和聚集的突变蛋白,导致常染色体显性家族性PD。突变的α-syn蛋白形成有毒的β-片状低聚物,导致线粒体功能障碍、氧化应激、钙稳态破坏和神经炎症,这些都导致神经变性。目前缺乏有效的预防α-syn聚集的治疗干预。
近年来的研究表明,肠道菌群可能在神经退行性疾病的发病机制中发挥重要作用。例如,抗生素治疗改善了PD小鼠的病理生理,治疗后的微生物再定殖恢复了PD症状。与移植健康供者的菌群相比,PD患者的α-syn过表达小鼠的定植加重了身体损害。除了动物模型,临床研究也为帕金森病中肠道和大脑的微生物联系提供了证据。胃肠功能障碍是PD患者的常见症状,幽门螺杆菌感染与疾病的严重程度和进展有关。对PD患者的粪便样本进行测序发现,与健康个体相比,肠道细菌组成发生了变化(例如,乳酸杆菌科增加。与PD相似,阿尔茨海默病(AD)小鼠模型中的肠道细菌促进了淀粉样蛋白病理(10),AD患者的肠道微生物组组成也发生了改变。
【6】Mol Cancer: 颅咽管瘤中CTNNB1新突变的全基因组序列分析
doi:10.1186/s12943-021-01468-7
颅咽管瘤(CP)是一种罕见的组织学良性肿瘤,但因其与中枢神经系统的重要结构密切相关,临床上极具挑战性。CP可分为两大亚型:造釉细胞型CP(ACP)和乳头型CP(PCP)。尽管在先前的研究中已经发现了这两类肿瘤的一些基因异常,但这种肿瘤的基因改变的完整谱仍不清楚。
CP是组织学上良性但具有临床挑战性的肿瘤,包括ACP和PCP亚型。最近,通过靶点测序或全外显子测序揭示了这两种cps亚型的分子特征。但是,这些测序技术可能会错过对一些重要基因组区域的关键洞察,而且CPS基因变化的完整谱仍然难以捉摸。
由于作者使用WGS分析了包括内含子和基因间隔区在内的整个染色体序列,作者的结果发现了更多的CP的遗传改变,并为CP的分子特征提供了更深刻的见解。例如,作者在的队列中发现了相对频繁的AHNAK突变(5/26)。AHNAK在多种信号通路中发挥重要作用,如转化生长因子β/smad信号转导,因此其表达失调被报道促进肿瘤的发生。
此外,ahnak突变被认为是与不同癌症患者生存不良有关的预后因素。在此,作者发现CPS在AHNAK基因中存在5个突变,其中1个是错义突变,4个是沉默突变,提示它们在CPS中具有重要意义。研究结果提供了一些有待解决的问题:这种错义突变是否改变了CPS中AHNAK的功能?这些突变是否可以作为CPS的预后标记物?为了了解突变对其功能的潜在影响,作者使用生物信息学工具来预测有害突变,然后分析它们在基因中的定位。有趣的是,作者发现38个预测的有害错义突变位于蛋白质的重要区域,这表明这样的突变可能会影响它们的蛋白质功能。TEC激酶是T细胞信号转导的重要组成部分,通过调节成纤维细胞生长因子-2的分泌调控人多能干细胞的早期细胞命运。
【7】PNAS:全基因组筛查肠道微生物在调节神经退行性疾病中的研究
doi:10.1073/pnas.2106504118
越来越多的证据表明,肠道微生物在调节神经退行性疾病(如帕金森病)的进展方面发挥着关键作用。这种微生物与宿主相互作用的分子机制还不清楚。在本研究中,作者通过用大肠杆菌敲除突变体喂养表达人α-syn的秀丽隐杆线虫,进行了全基因组筛选,以确定促进宿主神经变性的细菌基因。神经退行性疾病的特征是蛋白质错误折叠和聚集,导致β-片结构丰富的淀粉样原纤维的形成。这种蛋白质聚集引发蛋白质毒性,最终导致神经元死亡。DA神经元的缺失导致纹状体多巴胺信号的减少,从而导致PD患者运动功能受损。α-syn是一个小的(140个氨基酸)蛋白质,由一个n端结构域、一个非a -β组成的结构域(是纤维化核心)和一个羧基末端区域组成。n端结构域的错义突变,如A30P、G46K和A53T,通过产生比野生型更容易错误折叠和聚集的突变蛋白,导致常染色体显性家族性PD。突变的α-syn蛋白形成有毒的β-片状低聚物,导致线粒体功能障碍、氧化应激、钙稳态破坏和神经炎症,这些都导致神经变性。目前缺乏有效的预防α-syn聚集的治疗干预。
近年来的研究表明,肠道菌群可能在神经退行性疾病的发病机制中发挥重要作用。例如,抗生素治疗改善了PD小鼠的病理生理,治疗后的微生物再定殖恢复了PD症状。与移植健康供者的菌群相比,PD患者的α-syn过表达小鼠的定植加重了身体损害。除了动物模型,临床研究也为帕金森病中肠道和大脑的微生物联系提供了证据。胃肠功能障碍是PD患者的常见症状,幽门螺杆菌感染与疾病的严重程度和进展有关。对PD患者的粪便样本进行测序发现,与健康个体相比,肠道细菌组成发生了变化(例如,乳酸杆菌科增加。与PD相似,阿尔茨海默病(AD)小鼠模型中的肠道细菌促进了淀粉样蛋白病理(10),AD患者的肠道微生物组组成也发生了改变。
尽管紊乱的肠道微生物群和神经退行性疾病的发展之间的联系日益密切,但对细菌和神经系统之间通信的机械学理解是有限的。大多数理论关注的是全身炎症和异常微生物引起的神经炎症的神经退行性影响。细菌蛋白或代谢物是否能直接作用于宿主神经元,以模块由α-syn或A-β蛋白毒性引起的神经退行性变进程尚不清楚。这种局限性很大程度上是由于缺乏一个简单的模型来系统地测试单个细菌的成分对神经元的影响。

图片来源:Pixabay/CC0 Public Domain
【8】Science:对人类基因组和表观基因组的新见解将有助于预防、诊断和治疗癌症
doi:10.1126/science.abh1645
在2020年,估计有1000万人因癌症而丧生。这种毁灭性的疾病是由我们的DNA---我们所有细胞的指令手册---的变化所造成的。自从科学家们首次公布人类基因组序列以来,已经过去了20年。这一重大成就之后的重大技术进步,使我们今天能够非常详细地读取我们DNA的多层信息---从细胞癌变时发生的DNA的第一个变化到晚期肿瘤的复杂微环境。如今,为了加快对癌症患者的发现,我们需要新的方法来汇集我们产生的不同类型的复杂数据,以提供对癌症进化的生物学新见解。
如今,日本国家癌症中心研究所基因组学部主任Toshikazu Ushijima教授、新加坡基因组研究所执行主任Patrick Tan教授和澳大利亚加文医学研究所Susan J. Clark教授回顾了我们目前可以从分析DNA的全部复杂性中获得癌症新见解,并确定了我们需要解决的未来挑战,以便为患者带来下一步的变化。相关结果发表在2021年9月24日的Science期刊上,论文标题为“Mapping genomic and epigenomic evolution in cancer ecosystems”。
许多人把我们的DNA---我们的基因组---想象成简单的一串字母。在现实中,许多层信息---表观基因组---完全改变了它的活动。我们的基因组可以比作我们星球的不同地理环境。就像山脉、岛屿和海洋是由相同的基本元素组成的一样,我们的由碱基A、T、G和C组成的基因序列,构成了我们细胞内复杂结构特征的基础。这些地理环境是由我们的表观基因组---额外的信息层---形成的,包括附着在我们的DNA上的化学标记(称为DNA甲基化)和包裹着它的蛋白质(组蛋白)的化学变化,它们共同协调了DNA在我们细胞内的三维结构。
【9】Cell子刊: 人类诱导多能干细胞表观基因组异质性的内在基因组特性
doi:10.1016/j.celrep.2021.109909
人诱导多能干细胞(Human induced pluripotent stem cells, hipsCs)因其表观异质性而表现出不同的分化潜能,除了印迹和X染色体等研究充分的成分/染色体外,其分化程度/属性尚不清楚。在这里,作者展示了7个不同种系潜力的hipsC株系表现出明显的表观基因组异质性,尽管它们的转录组一致。
近四分之一的常染色体区域具有潜在的差异染色质修饰,H3K27me3/H2AK119ub1的启动子/CpG岛和H3K4me3的进化年轻的反转录转座子。作者鉴定了145个具有差异H3K9me3富集的大常染色体块(R100 kb),其中许多在体细胞中是层相关结构域(lad),而在胚胎干细胞中不是。这些表观基因组的大部分异质性与遗传变异无关。
多能干细胞(PSCs)包括胚胎干细胞(ESCs)和诱导型PSCs(ipsCs),具有强大的自我更新和分化为三层胚层细胞的能力。人类IPSCs(HipsCs)特别有利,因为它们是通过对分化的体细胞进行重新编程而产生的,因此绕过了使用人类胚胎引起的伦理问题。另一方面,hPSC的分化潜能表现出异质性。此前的研究表明,这种异质性与它们的遗传背景和表观基因组状态(包括DNA甲基化状态)的差异有关;然而,对这种异质性的分子基础的了解仍然有限。
与基因组印记和X染色体失活(XCI)相关的表观遗传异质性(我们使用的术语是指特定的区域/染色体)已经在人类esc (hESCs)和hipsCs中被描述。基因组印记通过亲本等位基因之间调控元件的不同DNA甲基化来调控特定基因(印迹基因)的亲本起源特异性表达;在hPSCs的印记区中,已经观察到亲代特异性DNA甲基化的缺失。XCI通过沉默女性的一条X染色体来补偿两性之间的剂量失衡。
【10】Science:人类着丝粒的完整基因组和表观遗传图谱
doi:10.1126/science.abl4178
为了在细胞分裂过程中忠实地将遗传物质分配给子细胞,纺锤丝必须通过一种称为“动粒(kinetochore)”的结构与DNA结合,动粒在每条染色体的着丝粒(centromere)上组装。人类着丝粒位于一大串称为α卫星重复序列(alpha satellite, αSat)的串联重复DNA序列内,这些序列通常横跨每条染色体上的数百万个碱基对。一大串αSat序列经常被其他类型的功能不甚明了的串联卫星重复序列所包围,同时还有包括可发生转录的基因在内的非重复序列。以前的基因组测序工作由于其规模和重复性,无法产生富含卫星重复序列的区域的完整组装,从而限制了研究它们的分布、变异和功能的能力。
在过去的20年里,人类参考基因组中几乎完全没有着丝粒周缘区域和着丝粒(pericentromeric and centromeric, peri/centromeric)卫星DNA序列。在一项新的研究中,来自端粒到端粒(Telomere-to-Telomere, T2T)联盟的研究人员使用完整的、端粒到端粒(T2T)的人类基因组组装,开发并部署了定制的计算方法,以揭示这些卫星重复序列在大的和小的长度尺度上的分化和进化模式。他们还进行了一些实验,以精确绘制哪些αSat重复序列与动粒蛋白相互作用。最后,他们比较了多个个体之间的peri/centromeric区域,以了解这些序列在不同的遗传背景下是如何变化的。相关研究结果发表在2022年4月1日的Science期刊上,论文标题为“Complete genomic and epigenetic maps of human centromeres”。
卫星重复序列占T2T-CHM13基因组的6.2%,其中αSat是最大的一个组成部分(占该基因组的2.8%)。通过对每个着丝粒的αSat的序列关系的详细研究,这些作者发现全基因组的证据表明人类着丝粒是通过“分层扩张(layered expansions)”进化的。具体来说,不同的重复序列变异出现在每个着丝粒区域内,并通过类似于连续串联重复(tandem duplication)的机制进行扩张,而较老的侧翼序列则随着时间的推移而缩小和分化。
这些作者还发现,每串αSat内最近扩张的重复序列更有可能与内部的动粒蛋白---着丝粒蛋白A(CENP-A)---相互作用,这与CpG甲基化降低的区域相吻合。这表明局部卫星重复序列扩张、动粒定位和DNA低甲基化之间有密切的关系。此外,他们发现了影响多种卫星重复序列类型的大型和意外的结构重排,包括活跃的着丝粒αSat。(生物谷Bioon.com)
生物谷更多精彩盘点!敬请期待!
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
