
Genome Medicine:浙江大学良渚实验室沈宁团队发布多维注释多类别变异致病性预测方法
来源:生物世界 2024-01-10 11:55
该研究建立了一个专注于对孟德尔遗传疾病的多种类型的未确定意义变异进行分类的算法框架。
良渚实验室沈宁研究员、伍赛教授,博士生刘逸程、张天韵等在 Genome Medicine 期刊发表了题为:MAGPIE: accurate pathogenic prediction for multiple variant types using machine learning approach 的研究论文。
该研究报道了使用多模态数据注释对多类型变异进行致病性预测的算法框架——MAGPIE(Multimodal Annotation Generated Pathogenic Impact Evaluator)。该算法是一个预测外显区域内除同义变异外其他类型变异的广泛适用模型,增强了致病性预测领域多类别变异整合预测的能力。将变异预测的结果与临床表型等结合能够在临床诊断、筛查疾病等方面具有重要价值。

由于导致遗传性疾病的不同种类致病变异通过多种生物学机制发挥作用,且算法需要面对多种不同的变异类型进行致病性的预测,研究团队开发了MAGPIE算法。MAGPIE算法主要分为三个阶段:多维特征注释,带有分离特征选择的自动特征工程,以及模型分布训练。
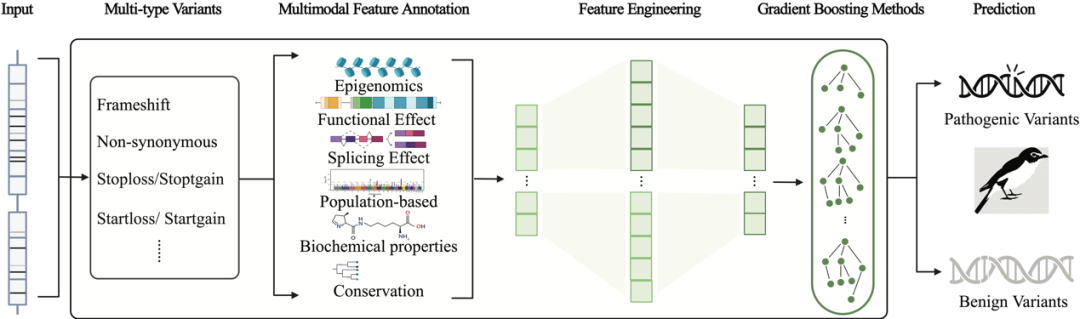
第一步将来自于ClinVar的经过验证的变异与来自gnomAD的良性罕见变异进行多维特征注释,特征类别包括:1)表观基因组学、2)功能效应、3)剪接效应、4)基于人群的特征、5)生化特性、6)保守性。这里加入gnomAD中罕见良性变异是为了消除原有ClinVar中数据中可能存在的bias,即数据中人群等位基因频率与变异致病性高度相关。
第二步,研究团队使用Oba等人提出的基于贝叶斯PCA的缺失值估计方法对经过注释的数据进行缺失值填补并通过一系列条件进行过滤筛选得到候选训练数据集,应用特征工程将特征数量扩充至3,000个以上。
为了避免过拟合问题,研究团队将所有特征分为两个类别,即核心集和附加集。前者包括与致病性直接相关的特征,如基于人口统计学的特征、保守性和功能效应。这些特征在训练阶段被全部保留下来。后者包括其他特征和自动特征工程生成的特征。研究团队将使用这些特征进行不超过50轮的训练,将特征减少到200个或更少。随后,在每一轮之后,研究人员评估每个特征的重要性,并丢弃相对重要性得分低于0.001的特征。最后,研究团队定义了五轮训练,每轮使用5折交叉验证,对于Light GBM中各参数进行分步调节,并在保证准确性和分类性能指标没有显著下降的条件下降低模型的过拟合问题,增强其泛化能力。
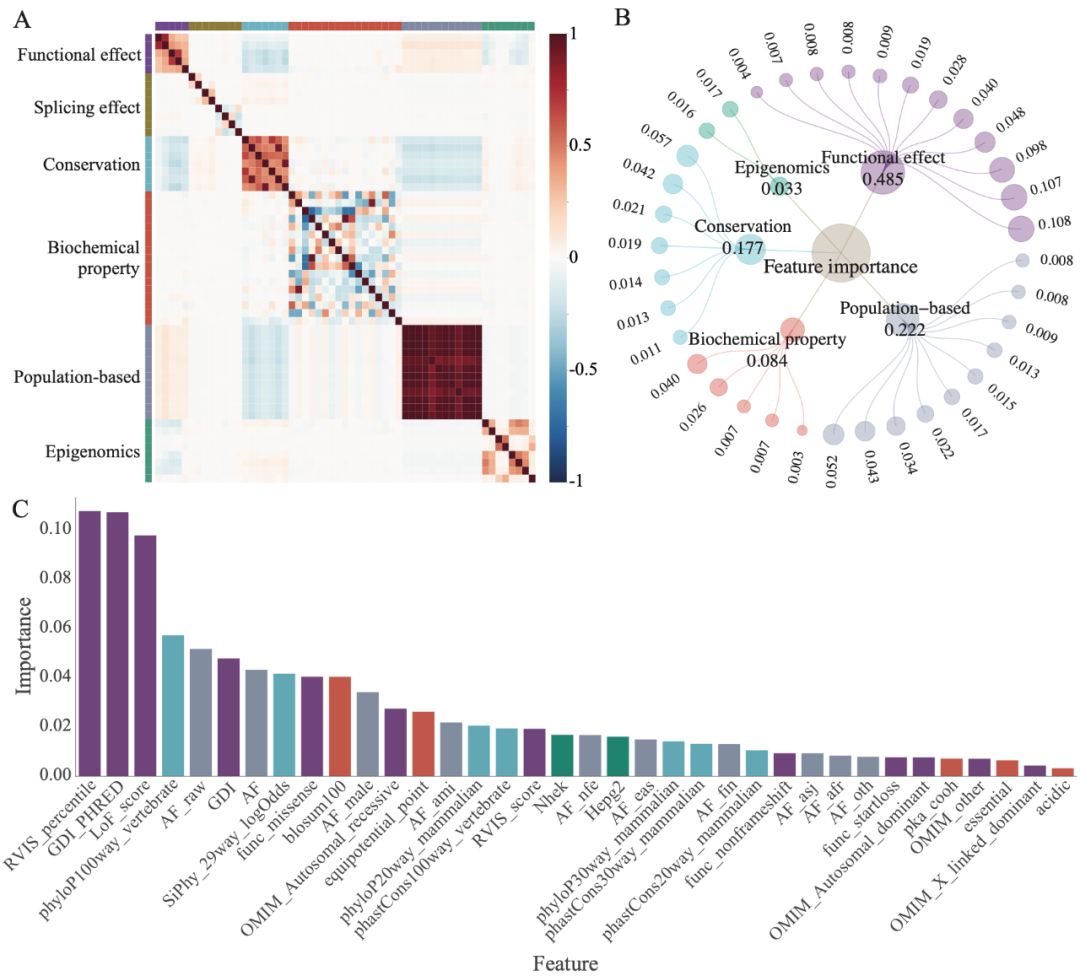
梯度提升方法的应用使得MAGPIE成为一个可解释的机器学习模型。为了更好地理解模型学习到的表示,研究团队计算了不同特征模态的重要性,并以网络视图的形式呈现。MAGPIE中的特征重要性表示每个特征对模型的贡献程度,并反映了它们的信息增益。在分类过程中,MAGPIE集成了六种特征模态。其中,最重要的特征组是功能效应,包括功能丧失得分(LoF_score)、人类基因损伤指数(GDI)等指标。之前研究结论为功能丧失变异更有可能导致疾病,与之相符的是研究人员发现MAGPIE预测的致病变异的LoF得分显著低于良性变异。GDI是每个编码蛋白质的人类基因的累积变异损伤,高度受损基因的变异更不可能导致疾病。研究团队观察到GDI特征被赋予了很高的权重,表明GDI较低的基因倾向于具有较高的MAGPIE预测分数,并更可能是致病的,与先前发表的研究一致。
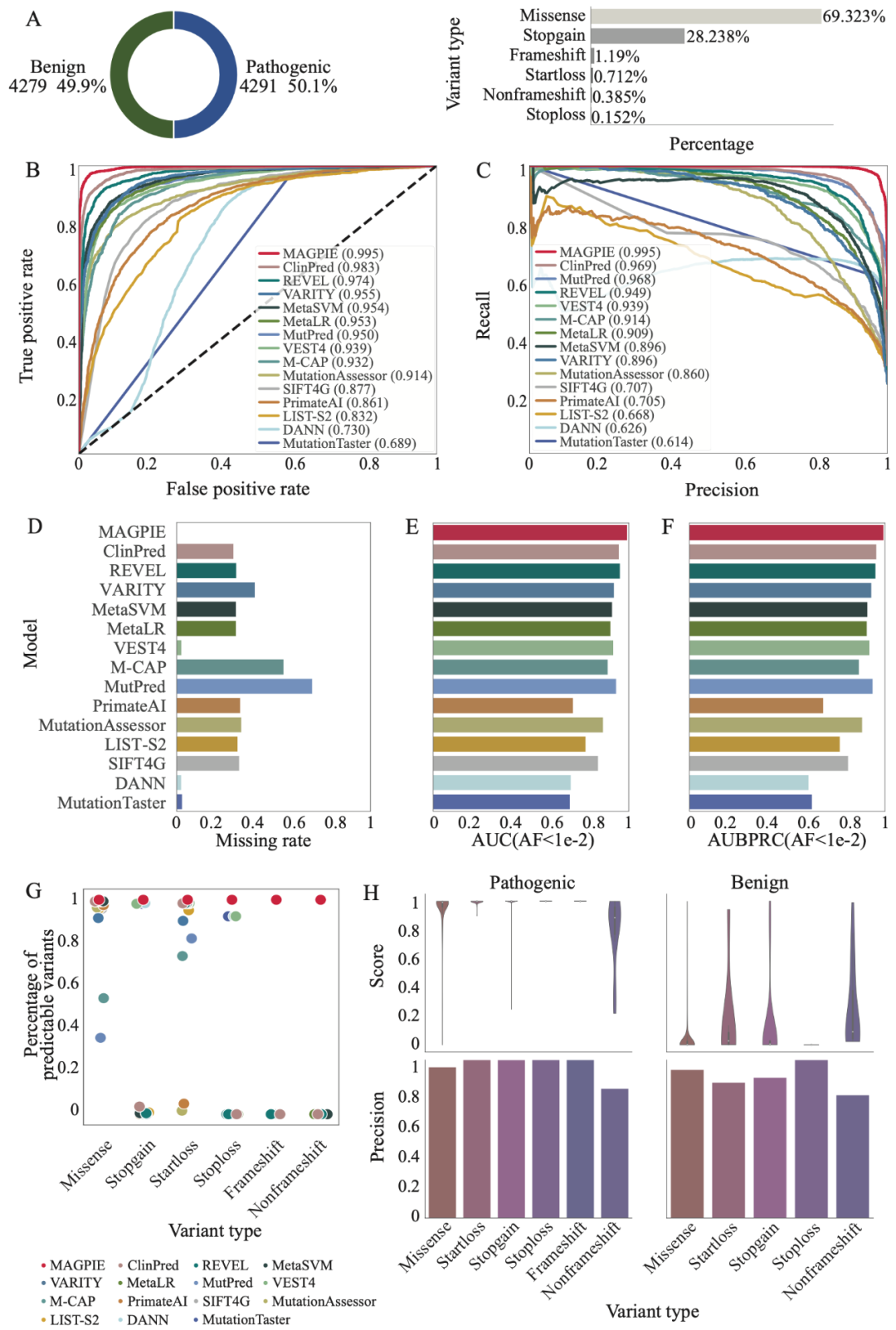
研究团队分别在三个不同的数据集,分别是ClinVar与gnomAD混合数据集independent test set、SwissProt与gnomAD混合数据集orthogonal validation set、由ACMG指导的临床验证数据集ACMG-guided dataset中对MAGPIE的性能进行了评估,其中independent test set为类别平衡数据集,即阳性样本与阴性样本数基本一致;orthogonal validation set中阳性样本占比超过90%,ACMG-guided dataset中阳性样本占比不足20%。研究人员发现MAGPIE可以在多种类别分布的数据集中稳定的实现0.95以上的AUC,0.88以上的AUPRC,以及0.9以上的准确率(accuracy)。MAGPIE同样在各数据集中准确预测所有变异类型,能够在移码变异(frameshift)、提前终止变异(stopgain)等变异类型中有均超过0.85的准确率。
为了检测MAGPIE在更符合真实临床数据下的性能表现,研究团队在上述三个数据集上筛选出罕见变异(AF<0.01)数据集并测试MAGPIE的性能。一般来讲,在罕见变异数据集上各方法的性能会略差于未经筛选的数据集,但相比于其他方法,MAGPIE在上述三个罕见变异数据集中拥有最小的性能削弱,并能够在各罕见变异数据集中达到0.9以上的AUC、0.85以上的AUPRC,与0.85以上的准确率。
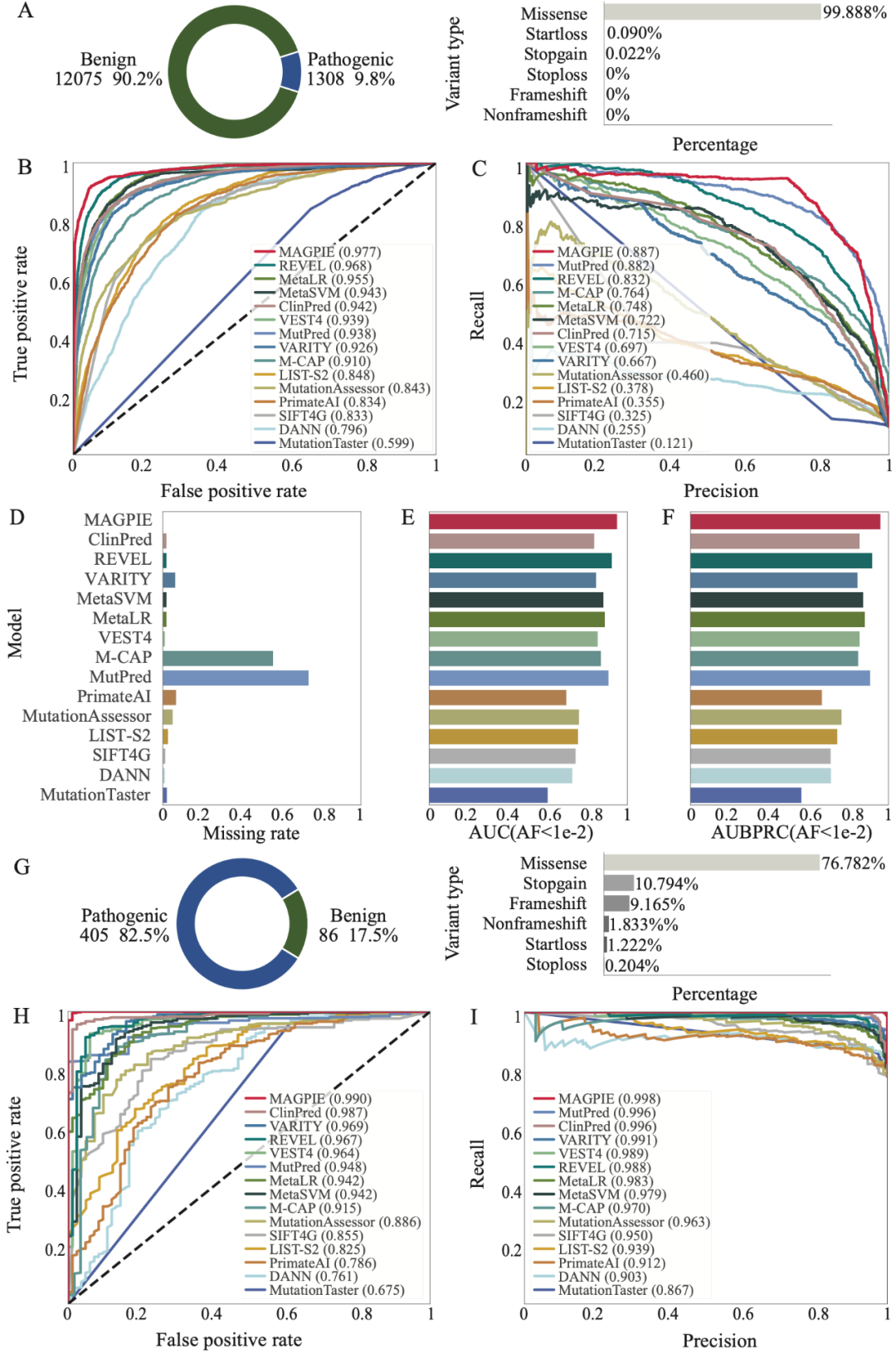
此外,研究团队为了进一步验证算法的性能,将MAGPIE应用于四个基因(ATP7B、CFTR、FBN1和LMNA),这些基因中包含大量已知经过实验验证的导致不同孟德尔遗传病的致病变异。对于上述基因,与其他方法的默认参数相比,MAGPIE能够识别出最多已知的致病变异,准确率平均在85%以上。例如,对于FBN1基因,该基因变异导致马凡综合征(Marfan syndrome),并包含1621个致病变异。MAGPIE成功预测出了1415(87%)个候选变异为致病。对于与囊性纤维化相关的CFTR基因,MAGPIE预测出了164(95%)个变异为致病。与MutationTaster、VEST4和其他工具相比,MAGPIE分数集中在1附近,这表明MAGPIE对致病变异的预测有更高的置信度。
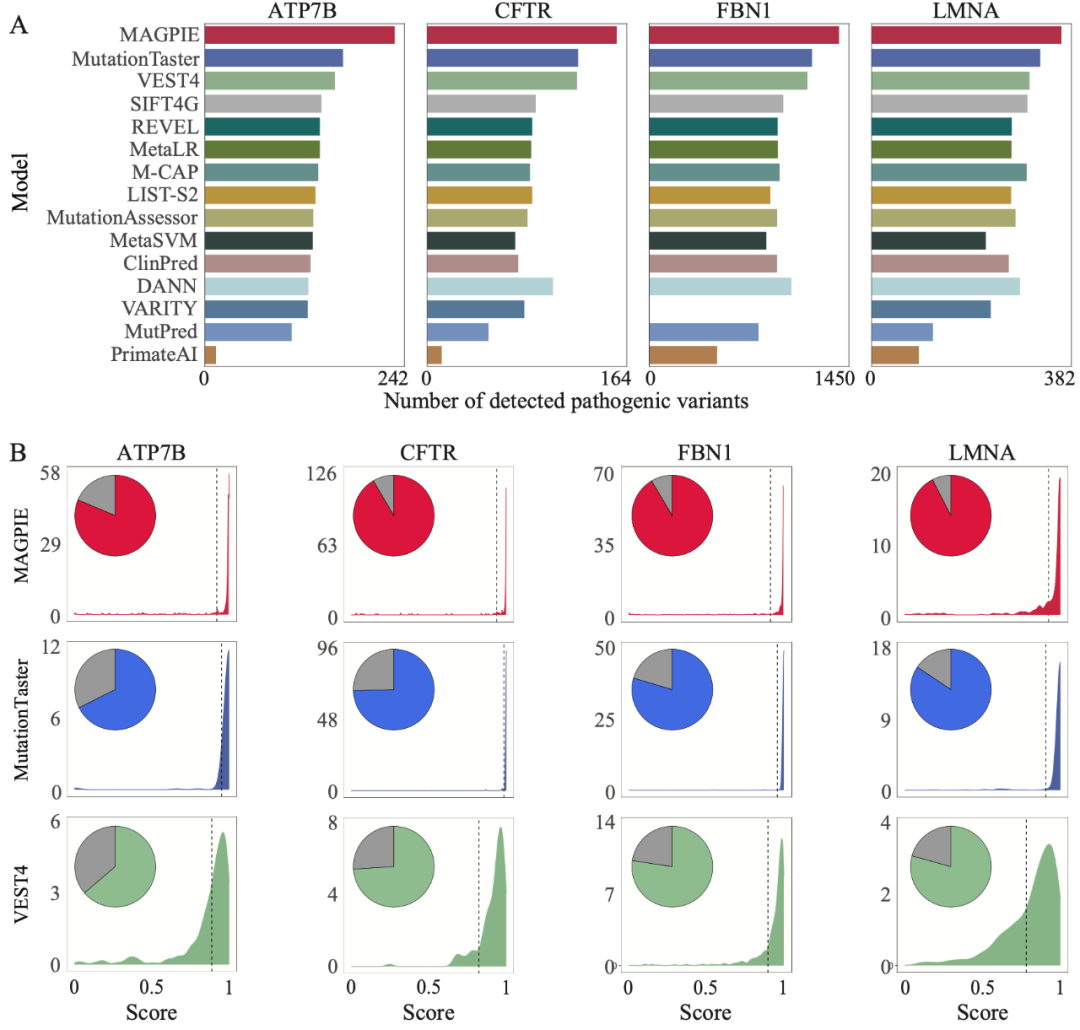
综上,该研究建立了一个专注于对孟德尔遗传疾病的多种类型的未确定意义变异进行分类的算法框架。这项工作整合了数据准备、特征提取、特征工程和模型选择。从用户的角度来看,变异的致病性应该在多个范围上进行联合评估,这对应于MAGPIE中的六种模态的特征。因此,MAGPIE的机器学习组件被专门设计,以适应多模态数据并能够最大化模型的学习能力。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
