
《科学》:重大突破!港中大杜洋团队等发现新型不成瘾的非阿片类止痛药
来源:奇点糕 2023-09-27 11:21
需要指出的是,尽管9087及其类似物的镇静副作用更低,但是其有效剂量要远高于dex,而且其作用机理和靶标也需要更深入的研究来解释。
近日,由蒙特利尔大学的Michel Bouvier,香港中文大学(深圳)的杜洋,加利福尼亚大学旧金山分校的Brian K. Shoichet和Allan I. Basbaum,以及埃朗根-纽伦堡大学的Peter Gmeiner联合领衔的研究团队,在《科学》期刊上发表了有关止痛药的重要研究成果[1]。
他们将目光聚焦于α2AAR,通过分子对接,从超过3亿个分子中,筛选出了若干个特异性靶向α2AAR的激动剂。在小鼠模型中,这些分子通过注射或者口服均能达到镇痛效果,且在有效剂量下没有镇静的副作用。

论文首页截图
接下来我们就一起来看看这个研究是如何展开的。
研究人员首先进行了分子对接实验。由于目前还没有α2AAR晶体结构的报道,研究人员使用了与α2AAR具有相似活性位点,且晶体结构已知的α2B-肾上腺素受体(α2BAR)。
他们将人α2BAR的晶体结构与3.01亿个分子进行对接试验,得到了64个评分高的结构,之后合成了其中的48个分子。其中,有30个分子与α2BAR的结合常数(Ki)都低于10uM,成功率达到63%,表现最好的分子ZINC1173879087(之后简写为9087),Ki达到1.7nM。
实验显示大部分分子都是α2BAR和α2AAR部分或者完全激动剂。其中有四个激动效果最好的分子,编号分别为:9087、4622、2998、0172。
生物发光共振能量转移(BRET)等实验表明,它们能激活α2AAR-Gi通路,效果可以达到去甲肾上腺素(NorEpi)的60-95%,EC50能达到9.7-210nM。α2AAR作为G蛋白偶联受体(GPCR),其激动剂既可能激活下游G蛋白的α亚基(Gi、Go或者Gz),从而发挥止痛功效,也有可能通过募集β-抑制蛋白,产生受体内化等作用。
9087以及其类似物7075、PS75均为Gi的部分激动剂,与实验中用到的NorEpi、dex、氯压定(clon)等已知的α2AAR的激动剂相比,筛选出的新分子大多能特异性激活Gi、Go和Gz通路,在有效浓度下,对β-抑制蛋白的募集作用可以忽略不计(0172除外),并且没有受体内化现象。

新激动剂(下)与已知激动剂(上)的的结构比较

Gi1激活(左)和β-抑制蛋白募集(右)实验
在筛选出以上四个最有潜力的激动剂分子(9087、4622、2998、0172)之后,研究人员使用冷冻电镜解析出了9087-α2AAR-GoA和4622-α2AAR-GoA的晶体结构,阐明了这些新配体如何与α2AAR相互作用。
尽管9087与dex和NorEpi一样都能与保守位点D128形成相互作用,但是分子朝向和作用方式非常不一样,而这也预示着9087及其类似物很可能是与现存激动剂作用方式不同的新型α2AAR激动剂。

9087-α2AAR-GoA的晶体结构
接下来,研究人员开展了基于9087的结构优化和构效关系分析。
他们发现,替换掉吡啶环、二级胺或者双环结构都会降低激动效果,他们还通过变换取代基获得了若干EC50更低的分子。

基于0987的结构优化和构效关系分析
随后研究人员进行了特异性和药代动力学测试。他们测试了320个GPCRs、µ-阿片受体、hERG(心脏安全相关基因)以及其他肾上腺素受体,9087均未表现出高亲和力。这证明9087的脱靶效应低,可能引发的副作用小。
值得注意的是,咪唑啉-2受体(I2R)是α2AAR激动剂的常见脱靶靶标,9087对α2AAR的亲和力是I2R的6倍,证明了其良好的特异性。之后他们进行了药代动力学测试,让人惊喜的是,无论通过注射或者口服,9087均能透过血脑屏障,在脑脊液中达到有效浓度。
在证明了新分子具有良好的特异性和生物利用度之后,研究人员在小鼠模型上测试了其止痛效果。在健康小鼠上使用9087(5mg/kg),没有增加机械缩足阈值,证明9087不会改变健康小鼠的疼痛阈值。
随后,他们给保留性神经损伤小鼠模型(SNI模型,小鼠由于部分周围神经损伤,对机械痛更加敏感)皮下注射9087,发现机械缩足阈值呈现剂量依赖性增加,从3mg/kg到5mg/kg有大幅度增加,之后便不再增加,处于平稳状态。
低剂量时(<5mg/kg),9087能减轻SNI小鼠对机械痛的敏感程度;高剂量(≥5mg/kg)时,9087的镇痛效果能将机械缩足阈值提高到比损伤前还要高的程度。
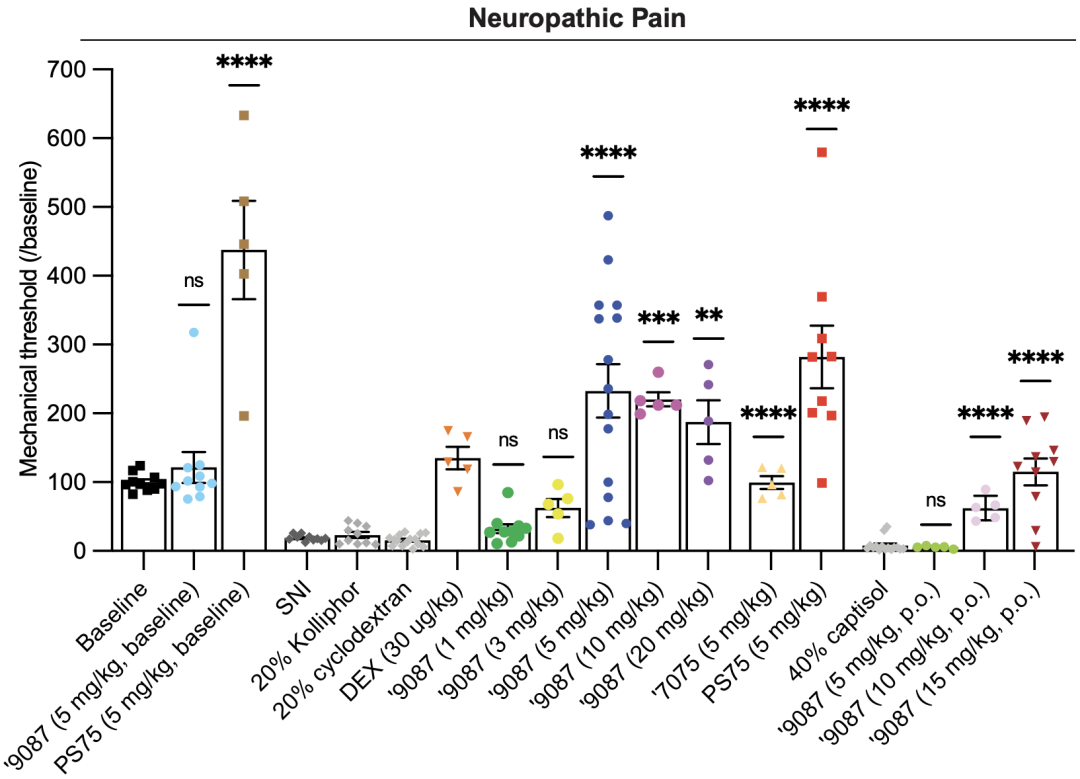
新激动剂在健康小鼠(左边3列)和SNI小鼠(右边15列)内对机械痛的作用效果
在完全弗氏佐剂(CFA)诱发的炎症疼痛小鼠模型中,9087可以提高热刺激缩足反应潜伏期。
以上两个实验证明,在小鼠模型中,9087可以缓解由组织和神经损伤导致的疼痛。
除此之外,他们还通过热板和尾闪实验,证明了9078可以缓解急性热刺激疼痛。
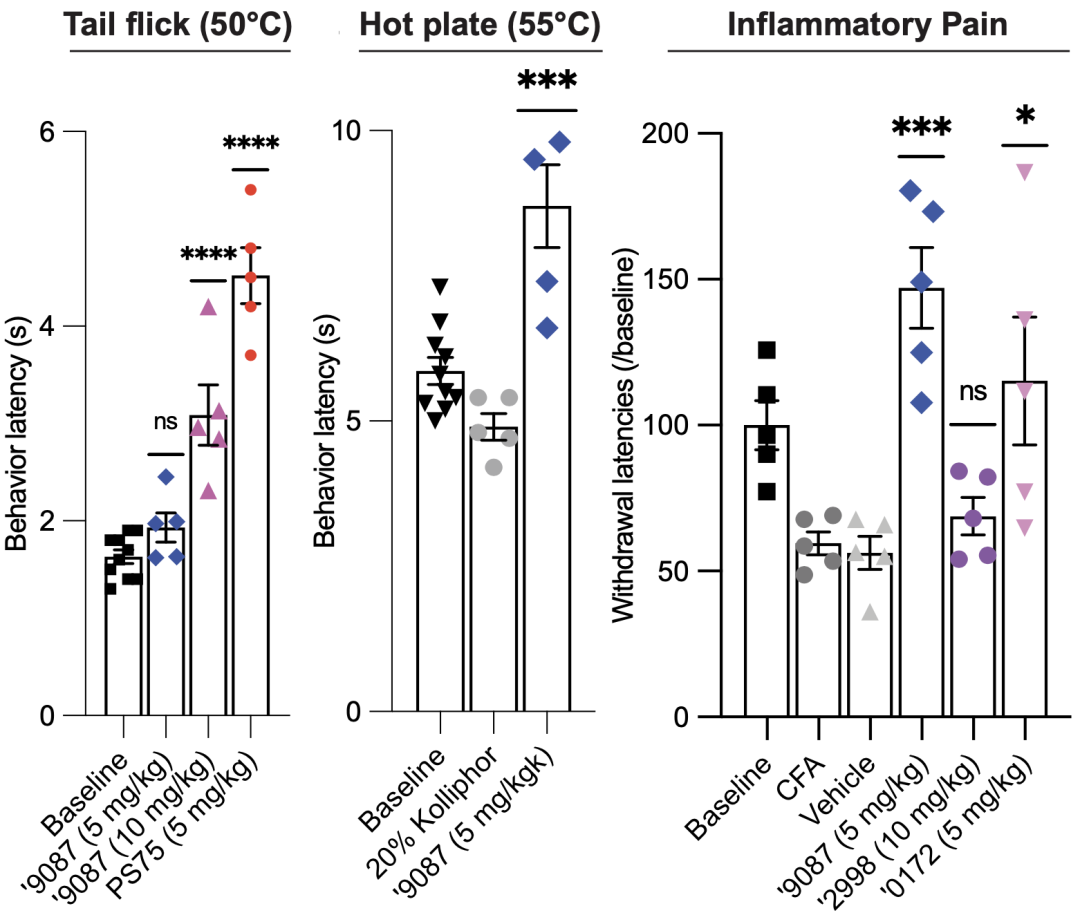
新配体在尾闪实验(左)、热板实验(中)、炎症疼痛模型(右)中的效果
为了验证9087是否有镇静的副作用,研究人员让使用高剂量9087的小鼠进行了滚轮跑步机实验,结果发现20mg/kg的9087也未能影响小鼠的运动表现。这证明9087不像dex一样有镇静的副作用。
接下来研究人员研究了9087镇痛的机理。
他们发现,使用广谱α2-肾上腺素受体(α2AR)的拮抗剂阿替美唑之后,9087就不再有止痛效果。然而,在使用点突变使α2AAR失活后(D79N),9087仍有镇痛效果,但是效果不到未突变组的50%。
这些实验结果表明,9087的镇痛效果虽然大部分是由于激活α2AAR引发的,但是可能也存在着其他靶标。
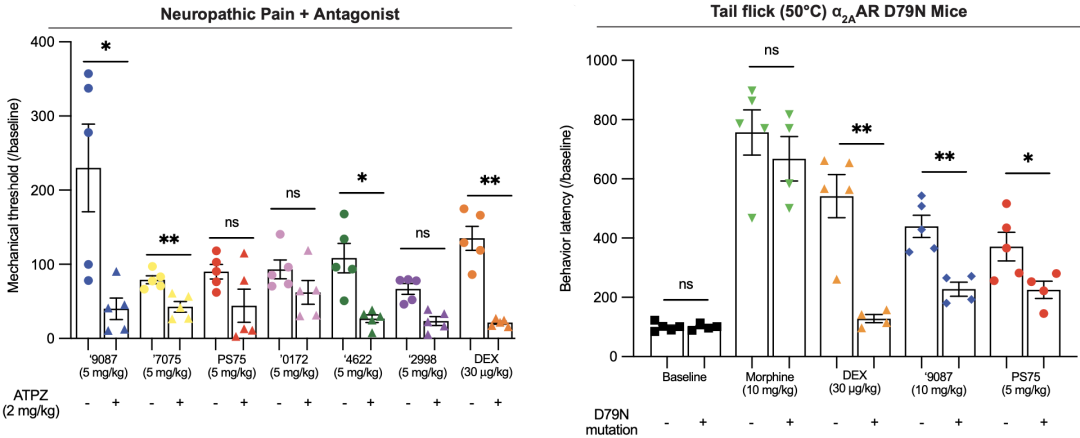
使用广谱拮抗剂后(左)和点突变使α2AAR失活后(右)新激动剂的镇痛效果
最后,研究人员测试了9087是否有止痛药的一些常见副作用。
好消息是,9087(10mg/kg)没有导致体重增加或是便秘等常见的α2AAR激动剂的副作用。除此之外,研究人员也测试了其他待选分子,它们虽然也有镇痛效果,但是整体来说还是9087的效果更好,副作用更低。
总的来说,这个研究通过大型分子库的对接试验,筛选出了一系列与已知α2AAR激动剂结构有较大差异的配体结构。这些分子特异性靶向Gi/o/z通路,其中分子9087及其类似物具有良好的口服生物利用度和镇痛效果,并且没有镇静的副作用,是非常有潜力的镇痛药候选分子。
需要指出的是,尽管9087及其类似物的镇静副作用更低,但是其有效剂量要远高于dex,而且其作用机理和靶标也需要更深入的研究来解释。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
