
《细胞》:中科院上海药物研究所团队揭示强效镇痛药芬太尼和吗啡的作用机理,有望让强效镇痛药摆脱成瘾性!
来源:奇点糕 2023-03-03 15:39
基于这些结论,研究者们成功设计了两个不与TM6/7互作的衍生物EBD1和EBD3。以芬太尼作为参考,两个衍生物对β-arrestin的激活均显著下降,并且保留了相似的激活Gi的能力。
吗啡和芬太尼均属于合成阿片类药物,在临床上被广泛地用于缓解疼痛。但是他们同时也具有严重的副作用,例如药物成瘾,呼吸抑制等等[1]。“阿片危机”,即指阿片类药物过量使用造成呼吸抑制进而导致死亡,是普遍存在各国的医疗问题[2]。因此,临床上亟需新型具有较少副作用的安全的镇痛药物。
为了筛选和设计无副作用的阿片类药物,首先要弄明白,现有的吗啡和芬太尼是如何引起副作用的。已有研究表明,它们的镇痛和副作用均主要通过激活μ型阿片受体 (μOR) 起效的[3]。
具体来看,μOR作为一种Gi偶联受体,由吗啡激活后启动细胞内信号通路,抑制下游的cAMP产生,进而产生镇痛作用。而呼吸抑制等副作用,是通过该受体介导的β-arrestin信号通路造成的[4]。在过去的几十年里,大量的工作都用来筛选μOR的具有较弱β-arrestin激活能力的激动剂。但是筛选出的药物,临床结果并不理想[5]。
因此,进一步弄清楚阿片类产生副作用的机理十分关键。而阿片类受体结构的研究无疑会带来有价值的细节信息。虽然已经有大量针对阿片类受体-配体结合复合物结构的研究,但是至今,仍然没有解出μOR与芬太尼结合的复合体的结构。
近日,中科院上海药物研究所徐华强/庄友文团队、谢欣团队和王明伟团队合作,在《细胞》上发表了重要研究成果。

文章首页
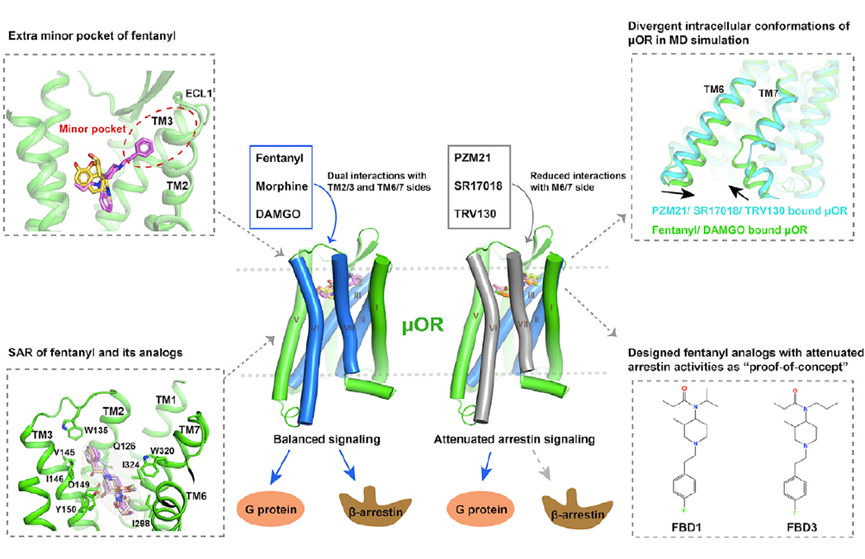
他们成功解析出了μOR分别与吗啡,芬太尼及其衍生物R17018、TRV130、PZM21结合复合体的高清冷冻电镜结构。这些结构阐释了吗啡、芬太尼与受体结合的特性,并且帮助研究者找出了激活β-arrestin信号通路的分子层面的决定性因素。他们进一步利用这些研究成果,成功设计了概念性的可激活Gi通路且无β-arrestin激活能力的芬太尼衍生物[6]。
该文章的图形摘要
首先,课题组利用单颗粒冷冻电子显微镜得到人源μOR与上述五种化合物分别结合的电镜结构,分辨率高达2.8至3.3 Å,并建立了μOR从S66到F352位氨基酸的模型。
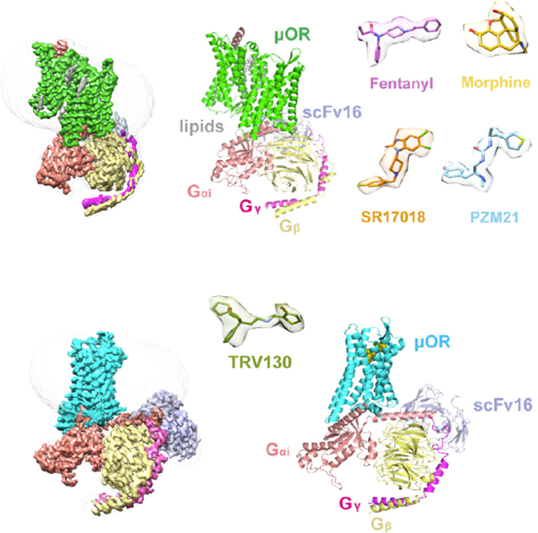
(上)芬太尼-μOR-Gi同型二聚体复合体的冷冻电镜密度图和分子模型。芬太尼,吗啡,SR17018和PZM21的密度图。(下)μOR-Gi与TRV130结合的结构
课题组解析到的高清结构图,展示了吗啡和芬太尼与μOR的特异互作方式,这也符合吗啡与芬太尼完全不同的化学结构。
进一步研究表明,当芬太尼、吗啡等其它激动剂与μOR结合后,会造成μOR一系列的结构改变,最终为G蛋白结合开放了胞内的腔室部分。
那么,这些μOR的配体是如何偏好性激活β-arrestin信号通路、引发呼吸抑制副作用的呢?
拟肽激动剂DAMGO,可以同时激活μOR的Gi和β-arrestin介导的下游通路。在这里,研究者们以DAMGO为阳性对照,检测发现,吗啡、芬太尼、R17018、TRV13和PZM21均可激活Gi通路从而抑制cAMP的生成。其中,PZM21不激活β-arrestin,TRV130和R17018部分激活β-arrestin,而芬太尼和吗啡强效激活β-arrestin。
随后,通过详细的对比以上这6个复合物的结构,研究人员发现,配体与μOR的第六(TM6)、第七(TM7)跨膜区域的稳定结合是激活β-arrestin的必要条件。当在TM6与TM7引入突变位点后,的确可以减弱β-arrestin的激活。
为进一步探究激动剂造成μOR胞内构变与Gi /β-arrestin激活偏好的关系,研究人员进行了μOR与R17018、TRV13和PZM21互作的分子动力学模拟,并且比较了其他具有招募β-arrestin能力的G蛋白偶联受体,如M2受体的结构。
研究人员发现,当μOR与吗啡和芬太尼结合时,TM6和TM7向跨膜中心移动,造成受体的胞内可与G蛋白结合部分受挤压而减小,而可与β-arrestin的结合面积变大。而R17018、TRV13和PZM21与μOR结合时,TM6和TM7-H8远离TM中心,从而降低β-arrestin的结合能力。
令人欣喜的是,基于这些结论,研究者们成功设计了两个不与TM6/7互作的衍生物EBD1和EBD3。以芬太尼作为参考,两个衍生物对β-arrestin的激活均显著下降,并且保留了相似的激活Gi的能力。
该研究具有非常重要的理论和临床价值。通过解析μOR与芬太尼结合的结构,研究团队揭示了芬太尼特异结合模式,为研究众多的芬太尼衍生物的构效关系提供了合理的模型。不仅如此,他们阐释了μOR招募β-arrestin的结构细节,为将来设计更安全更好的镇痛类药物提供了模板。
参考文献:
1.Manglik A. Molecular Basis of Opioid Action: From Structures to New Leads. Biol Psychiatry. 2020;87(1):6-14.
2.Mattson CL, Tanz LJ, Quinn K, Kariisa M, Patel P, Davis NL. Trends and Geographic Patterns in Drug and Synthetic Opioid Overdose Deaths - United States, 2013-2019. MMWR Morb Mortal Wkly Rep. 2021;70(6):202-207.
3.Matthes HW, Maldonado R, Simonin F, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383(6603):819-823.
4.Raehal KM, Walker JK, Bohn LM. Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314(3):1195-1201.
5.Viscusi ER, Skobieranda F, Soergel DG, Cook E, Burt DA, Singla N. APOLLO-1: a randomized placebo and active-controlled phase III study investigating oliceridine (TRV130), a G protein-biased ligand at the µ-opioid receptor, for management of moderate-to-severe acute pain following bunionectomy. J Pain Res. 2019;12:927-943.
6.Zhuang Y, Wang Y, He B, et al. Molecular recognition of morphine and fentanyl by the human μ-opioid receptor. Cell. 2022;185(23):4361-4375.e4319.
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
