
Nat Cell Biol | β细胞保护机制的突破:m^6A甲基化在1型糖尿病中的关键作用
来源:生物世界 2024-03-03 10:00
该研究报告了m6A在人类T1D发病期间在β细胞水平调节先天免疫反应。
芝加哥大学何川及哈佛医学院Rohit N. Kulkarni共同通讯在Nature Cell Biology 在线发表题为“Redox regulation of m6A methyltransferase METTL3 in β-cells controls the innate immune response in type 1 diabetes”的研究论文,该研究发现N6 -甲基腺苷(m6A)是一种适应性β-细胞保护机制,在T1D发病时控制抗病毒先天免疫反应的幅度和持续时间。m6A“书写者”甲基转移酶3 (METTL3)水平在T1D发病时在β-细胞中急剧升高,但随着疾病进展迅速下降。m6A测序结果显示,在T1D发病时,人胰岛和EndoC-βH1细胞中几种关键的先天免疫介质(包括OAS1、OAS2、OAS3和ADAR1)的m6A高甲基化。
METTL3沉默通过增加2 ' -5 ' -寡聚腺苷酸合成酶mRNA的稳定性来提高其水平。与此一致的是,在非肥胖糖尿病小鼠T1D模型中,延长β细胞中Mettl3过表达的体内基因治疗延迟了糖尿病的进展。从机制上讲,活性氧的积累阻止了METTL3响应细胞因子的上调,而生理水平的一氧化氮增强了METTL3的水平和活性。此外,还报道了METTL3蛋白锌指结构域C276和C326位置的半胱氨酸对S-亚硝基化敏感,并且对METTL3介导的人β细胞低聚腺苷酸合成酶mRNA稳定性的调节很重要。总之,该研究报告了m6A在人类T1D发病期间在β细胞水平调节先天免疫反应。

区分自我与非自我DNA或RNA的能力是先天免疫系统的一项基本功能。因此,一些自身免疫性疾病是由先天免疫系统的过度激活引发的。此外,大量证据表明,在1型糖尿病(T1D)发病前,介导先天免疫系统的多个基因被激活。这一点具有重要意义,因为参与先天免疫反应的核酸传感器在T1D发病时在胰岛素胰岛中上调。上调的基因包括2 ' -5 ' -寡腺苷酸合成酶(OAS)家族,这是一类核苷酸转移酶,一旦被激活,要么独立起作用,要么产生2 ' -5 '连接的寡腺苷酸来激活RNase L。
值得注意的是,OAS基因簇的多态性与T1D的易感性有关。有趣的是,β-细胞在胰岛细胞中是独一无二的,它具有上调OAS表达的能力到干扰素(IFN)-α或聚(I:C)(双链RNA (dsRNA)模拟物)。在dsRNA诱导的T1D小鼠模型中,OAS在β-细胞中的过度表达导致增殖阻滞和凋亡,而RNase L缺失的小鼠可免受糖尿病的影响,这与OAS-RNase L通路的过度激活导致β-细胞死亡和T1D14的观点一致。
m6A是信使RNA中最丰富的修饰。腺苷甲基化水平由甲基转移酶3 (METTL3)和14 (METTL14)等“书写”蛋白调节。几种RNA结合蛋白,“阅读器”,包括YT521-B同源家族蛋白(例如,YTHDF),识别甲基化腺苷并调节mRNA生物学的几个方面,包括mRNA衰变。METTL3是m6 A书写复合物中唯一具有催化活性的酶。最近的研究表明,METTL3的活性可以通过SUMOylation和磷酸化来调节。然而,半胱氨酸氧化修饰如S-亚硝基化(SNO)的作用尚未被探索。
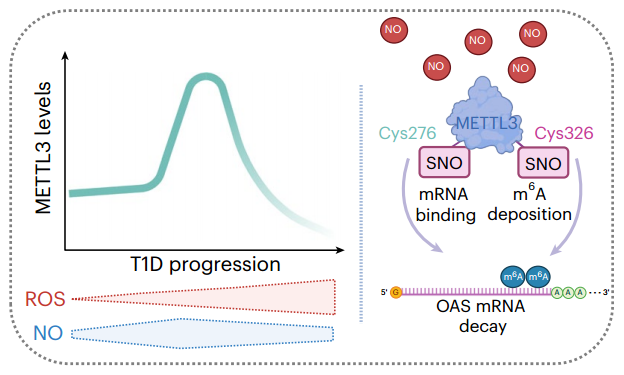
SNO在控制METTL3功能和OAS mRNA衰变中的作用(Credit: Nature Cell Biology)
最近的发现表明,m6A机制通过促进腺苷向肌苷(A-to-I) RNA编辑调节ADAR1或阻断内源性异常dsRNAs的合成的转变,加速成纤维细胞中I型IFN基因的周转,从而调节先天免疫反应。然而,m6A在T1D中的生物学作用,更具体地说,它们对介导β细胞先天免疫反应的贡献尚不清楚。
该研究发现METTL3水平在T1D发病时升高,然后迅速下降。此外,OAS基因的m6A高甲基化,并证明了METTL3在人pseudoislets 和EndoC-βH1细胞中的下调会导致OAS蛋白的上调。还观察到,m6A通过氧化还原敏感的METTL3锌指结构域的半胱氨酸残基(C276和C326)的SNO加速了OAS mRNA的衰变。β细胞中Mettl3的持续过表达能够限制Oas的上调,并保护非肥胖糖尿病(NOD)小鼠模型的T1D不发生糖尿病,这支持了这些发现的翻译意义。该研究确定了m6A作为一种适应性β细胞保护机制,在T1D发病时控制先天抗病毒免疫反应。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
