
MBD2缺乏通过miR-345-5p/atf1轴减轻高糖损伤和链脲佐菌素诱导的糖尿病小鼠视网膜细胞凋亡
来源:本站原创 2021-12-30 10:46
糖尿病视网膜病变(DR)仍然是劳动年龄人群致盲的主要原因。目前,视网膜病变的临床特征是明显的血管异常,而且微血管并发症可引发不可逆的视网膜损伤。
糖尿病视网膜病变(DR)仍然是劳动年龄人群致盲的主要原因。目前,视网膜病变的临床特征是明显的血管异常,而且微血管并发症可引发不可逆的视网膜损伤。DNA甲基化被认为在糖尿病视网膜病变的发生发展中起重要作用。在此,本研究的目标是研究甲基-CpG结合域蛋白2(Mbd2)在糖尿病早期视网膜神经节细胞(RGCs)凋亡中的确切作用。

图片来源: https://doi.org/10.1016/j.omtn.2021.10.026.
首先,作者评估了HG是否能诱导Mbd2的表达。Western blotting分析显示,在HG刺激后24h和72h,原代RGCs中Mbd2的表达水平明显升高。如前所述,mbd2已被报道在脑缺血后的脑组织中高表达,并在大鼠海马中诱导表达。活体免疫荧光图像还显示,STZ诱导的糖尿病后12周,Mbd2在RGC层的表达水平增加。
有趣的是,在视网膜的内核层和外核层也观察到了这种变化,这意味着Mbd2在其他视网膜细胞中的功能可能需要进一步研究。最近的一项研究表明,在万古霉素引起的急性肾损伤中,mbd2基因的缺失改善了人肾小管上皮细胞的凋亡。在本研究中,作者证明了Mbd2siRNA在体外和Mbd2基因敲除小鼠体内都能改善HG条件下RGCs的凋亡。这些数据表明,Mbd2至少在一定程度上介导了糖尿病视网膜神经细胞凋亡的进程。
接下来,作者研究了Mbd2在HG诱导的视网膜细胞凋亡发病机制中的作用机制。有趣的是,非编码RNA,特别是miRNAs,显示出更广泛的与表观遗传调控的联系,许多miRNAs已知受到表观遗传修饰的调控,包括DNA甲基化。Mbd2是一种甲基化的CpG结合蛋白。最近的一项研究报道miR-301a-5p在万古霉素诱导的急性肾损伤中受表观遗传机制的调控,Mbd2与miR-301a-5p启动子区域的甲基化CpG结合,从而抑制其甲基化。
作者还对参与细胞凋亡过程的miR-345-5p的靶基因进行了研究。已知miRNAs通过与靶mRNA的30UTR结合,在转录后调节基因表达。基于miRBase,来自先前研究的数据,作者关注atf1,它激活基因,包括braf,nras,myc,birc2,daam1,maml2,stat1,id1和nkd2,与细胞凋亡,wnt,TGF-b和MAPK通路相关,这些作用协同增加了结直肠癌的风险。
Sirna缺失atf1基因可显著加重hg诱导的视网膜节细胞凋亡,并使裂解的caspase3表达水平增加,表明atf1是一种抗凋亡因子。这些结果表明miR-345-5p在HG损伤后视网膜细胞死亡的发病机制中以抗凋亡基因atf1为靶点。有趣的是,Mbd2 siRNA过表达miR-345-5p可以部分逆转HG诱导的RGCs凋亡水平的降低。
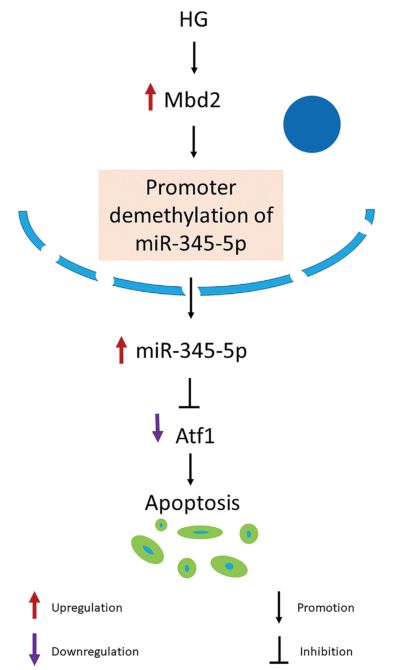
Mbd2通过miR-345-5p/atf1轴调控HG诱导的视网膜节细胞凋亡机制示意图
图片来源: https://doi.org/10.1016/j.omtn.2021.10.026.
在目前的研究中,作者直接研究了Mbd2在HG和糖尿病视网膜中RGCs凋亡机制中的作用。本研究首次报道了Mbd2介导HG诱导的视网膜节细胞凋亡。在体内,Mbd2基因敲除小鼠显著降低stz诱导的RGCs凋亡;这伴随着视觉功能的部分改善。机制上,作者发现Mbd2通过抑制启动子甲基化直接上调miR345-5p的表达,从而导致HG诱导的视网膜神经细胞凋亡。(生物谷 Bioon.com)
参考文献
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
