
Cell Discov | 深度学习模型可提高碱基编辑结果的预测准确性
来源:生物探索 2024-02-26 19:41
该研究开发了一种深度学习算法,用于准确预测生物多样性编辑结果。
中国科学院脑科学与智能技术卓越创新中心孙怡迪、中国农业科学院左二伟及中国科学院上海营养与健康研究所Wei Wu共同通讯在Cell Discovery(IF 34)在线发表题为“Deep learning models incorporating endogenous factors beyond DNA sequences improve the prediction accuracy of base editing outcomes”的研究论文,该研究表明结合DNA序列以外的内源性因素的深度学习模型提高了碱基编辑结果的预测准确性。该研究在哺乳动物细胞中对5012个内源性基因组位点和11868个基因组整合靶序列进行了ABE和CBE的碱基编辑结果评估,其中4654个基因组位点共享相同的靶序列。
比较分析显示,ABE和CBE在内源性位点的编辑结果与使用基因组整合序列获得的结果有很大不同。研究发现ABE和CBE在内源性靶点的碱基编辑效率都受到内源性因素的影响,包括表观遗传修饰和转录活性。基于基因组数据集的内源性因素和序列信息,开发了一种名为BE_Endo的深度学习算法,在预测碱基编辑结果方面取得了前所未有的准确性。这些发现以及开发的计算算法可能有助于未来生物等效体在科学研究和临床基因治疗中的应用。

单核苷酸变异(SNVs)占人类基因组致病性突变的一半以上,而SNVs的准确逆转是基因治疗的最重要目标之一。碱基编辑器(BEs),包括ABEs和CBEs,已被广泛用于纠正致病性点突变和生成动物疾病模型。然而,编辑结果的实验评估是耗时的,这限制了它的应用仅限于少数目标位点。最近已经开发了几种计算方法,利用哺乳动物细胞中慢病毒整合文库的靶向序列信息来预测BEs的编辑结果。
慢病毒整合文库通常包含数千个寡核苷酸,每个寡核苷酸编码一个独特的20个核苷酸(nt)的小向导RNA (sgRNA)间隔序列,并具有成对的靶序列。sgRNA文库随机整合到哺乳动物细胞基因组中,sgRNA在人类U6启动子的驱动下表达。表达的sgRNA与转染的或基因组整合的BEs结合,诱导碱基编辑综合目标序列。然后对整合的成对目标序列进行PCR扩增,并进行测序以测量编辑效率。
以往的研究表明,转录活性、染色质可及性等内源性因素与CRISPR-Cas9内切酶的切割效率密切相关。慢病毒整合文库限制了内源性因子在靶位点的检测,因为靶序列是随机整合到基因组中的。因此,需要生成大的全基因组内源性数据集来阐明内源性因素对碱基编辑的影响。然后可以开发包含非常重要的内源性因素的计算方法,以便更好地预测内源性碱基编辑的结果。
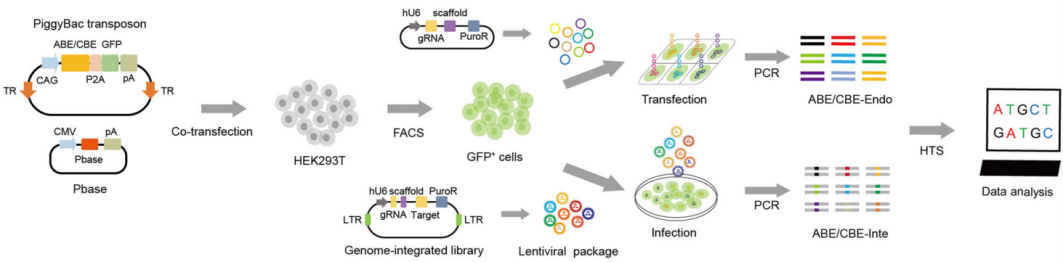
全基因组内源性和集成ABE和CBE数据集的生成概述(Credit: Cell Discovery)
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
