
鉴别全球各大洲人群中干扰群体遗传结构推断的基因组区域
来源:网络 2022-11-23 19:24
自人类的祖先走出非洲后,人类经历的众多迁徙,基因混合以及自然选择等历史事件在基因组中留下了印记,形成了群体遗传结构。
自人类的祖先走出非洲后,人类经历的众多迁徙,基因混合以及自然选择等历史事件在基因组中留下了印记,形成了群体遗传结构。因此,推断群体遗传结构对于研究人群的演化历史至关重要,还能帮助校正全基因组关联分析(GWAS)中人群结构的混杂效应。2008年,Price等人发现了欧洲人群基因组中的一些区域会干扰人群群体遗传结构推断。但是,不同大洲的人群拥有不同的演化历史,并且近年来基因检测技术和人群基因组数据日新月异,亟需对这一问题进行系统性分析和更新。
近日,华中科技大学公共卫生学院王超龙教授团队基于千人基因组计划(1000 Genome Project)GRCh38版本人类参考基因组的高深度(30×)全基因组测序数据,通过系统分析,在欧洲,东亚,南亚,非洲人群中共找到27个干扰群体遗传结构推断的区域。在对人群进行主成分分析(PCA),或用ADMIXTURE软件推断群体遗传结构时,排除这27个基因组区域,可得到更准确的推断结果。相关文章“Identification of genomic regions distorting population structure inference in diverse continental groups”(点击文末“阅读原文”下载PDF全文)发表在Quantitative Biology期刊上。

全文概要
作者把对推断群体遗传结构具有异常贡献的基因组区域定义为“不寻常区域”。作者创建了一套生物信息分析流程用以确定这些“不寻常区域”,并分别应用于四个大洲的人群数据。
首先,对质控后的基因型数据进行PCA推断群体遗传结构,并得到每个变异位点对推断结果影响的大小,并据此确定对推断结果有异常影响的基因组区域。在去掉这些区域后,并基于剩余区域的基因型再次进行PCA。这个过程重复迭代多次,直到下一轮PCA不再出现新的异常区域。我们将每次迭代所找到的区域综合到一起,即为该大洲人群最终的“不寻常区域”(见图1)。作者在欧洲人群中发现了17个此类区域,东亚13个,南亚10个,非洲5个。随着大洲内部的遗传多样性增高,此类区域的数量减少。
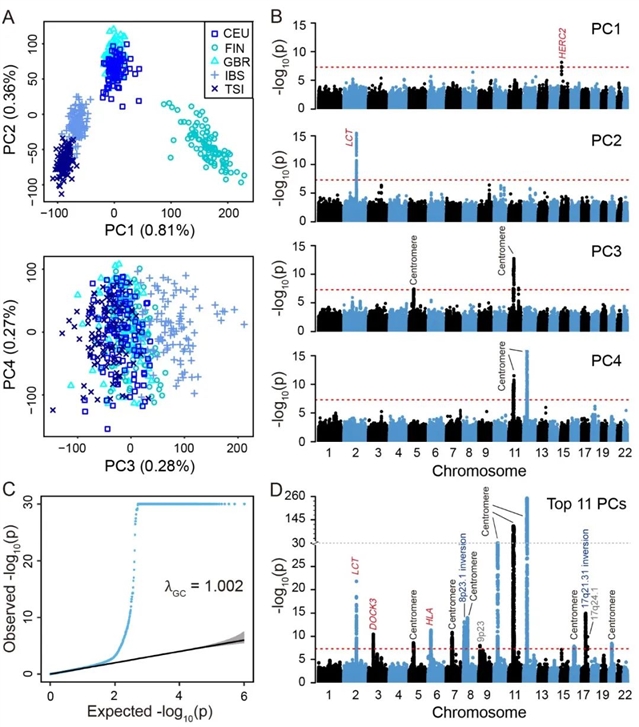
图1. 欧洲人群中的“不寻常区域”
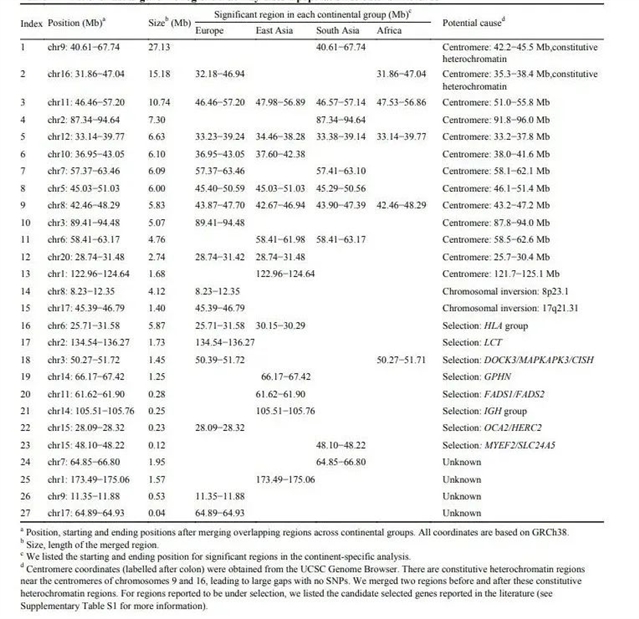
作者对27个“不寻常区域”按其可能产生原因分为四类:区域位于染色体中心粒附近(13个),区域经历了染色体倒位(2个),区域受到自然选择(8个)和原因不明(4个)(见表1)。中心粒附近的区域由于对细胞的正常分裂有着至关重要的作用,其重组发生率很低,导致有着很长的连锁不平衡(LD)区域。另外染色体倒位和自然选择均可导致强LD。
表1. 影响群体遗传结构推断的基因组区域(unusual regions)
接着,作者通过比较去除“不寻常区域”前后ADMIXTURE分析的结果发现,欧洲人群中的群体遗传结构推断受到的影响最大,去除后的结果更符合欧洲人的演化历史(见图2)。这也表明在遗传多样性较低的人群中,这些区域对群体遗传结构推断的结果产生的影响更为明显,与相关研究的结果一致。
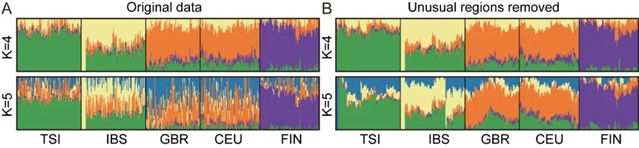
图2. 欧洲人群中unusual regions对ADMIXTURE结果的影响
最后,作者建议在使用人类基因型数据推断群体遗传结构时,应首先排除本研究中鉴定出来的27个“不寻常区域”中的遗传变异位点,以获得准确的群体遗传结构推断,有利于进行下游的演化历史分析或全基因组关联分析。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
