
KAI1(CD82)是控制血管生成和将血管生成环境切换到静止状态的关键分子
来源:本站原创 2021-10-21 15:47
关于血管生成生长因子的内源性抑制剂知之甚少。在本研究中,作者发现了一种新的内源性抗血管生成因子在周细胞中表达,并阐明了其潜在的机制和临床意义。
关于血管生成生长因子的内源性抑制剂知之甚少。在本研究中,作者发现了一种新的内源性抗血管生成因子在周细胞中表达,并阐明了其潜在的机制和临床意义。在此,作者发现Kai1基因敲除小鼠的血管生成明显增强。然后,作者研究了Kai1体外和体内抗血管新生roll的作用。
KAI1主要表达于周细胞而非内皮细胞。经zDHHC4酶棕榈酰化后定位于膜表面,通过Src/p53途径诱导LIF。周细胞释放的LIF反过来抑制内皮细胞和周细胞本身的血管生成因子,导致血管生成的抑制。有趣的是,KAI1还有另一种抑制血管生成的机制:它直接与VEGF和PDGF结合,抑制其受体的激活。在两种不同的体内肿瘤模型中,添加KAI1显著抑制肿瘤血管生成和生长。来自KAI1大细胞外环的一种肽已被证明具有抗血管生成作用,可在体内阻止乳腺癌的进展和视网膜新生血管。

图片来源:https://doi.org/10.1186/s13045-021-01147-6
血管生成是一个从现有的血管系统中形成新的血管的过程。这一过程主要受两种血管细胞的相互作用控制,内皮细胞(ECs)和周细胞(PCs)。尽管内皮细胞在血管生成中的作用已经被广泛研究,但除了血管内稳定和旁分泌信号外,内皮细胞在血管生成中的作用仍不清楚。此前,作者发现KAI1是一个缺氧相关基因,在缺血心肌和缺氧骨髓干细胞生态位中表达。
KAI1/CD82 是一种跨膜蛋白,是四跨膜蛋白超家族的成员,是一种在各种组织类型中表达的进化保守分子。KAI1首次被发现参与T细胞激活过程,通常被认为是一种转移抑制因子。KAI1主要在癌细胞和内皮细胞中抑制转移和血管生成。最近,作者等人报道了KAI1调控长期再生造血干细胞(long- repopulating hematopoietic stem cells, ht - hscs)和肌肉干细胞祖细胞的细胞周期进程。因此,KAI1在每个细胞和器官类型中都有不同的作用。然而,KAI1在血管周细胞/周细胞中的具体作用尚不清楚。
通过维持促血管生成和抗血管生成因子之间的适当平衡,血管生成受到严格控制。大多数用于癌症或血管增生的抗血管生成药物会阻碍血管内皮生长因子(VEGF)的信号传导。VEGF引起血管生成的机制是众所周知的,但内源性VEGF抑制剂及其抗血管生成过程的控制机制知之甚少。到目前为止,至少有27种内源性血管生成抑制剂已经被鉴定和检测到血液中。大多数来源于大的细胞外基质和非基质来源的分子碎片; 然而,tetrasppanin超家族衍生的内源性血管生成抑制剂尚未见报道。在这里,作者发现,在PCs中表达的KAI1是一种重要的内源性反调节因子,可抑制生长因子(如VEGF和PDGF)驱动的血管生成,而血管生成的稳态是由PCs和ECs之间的旁分泌相互作用控制的。
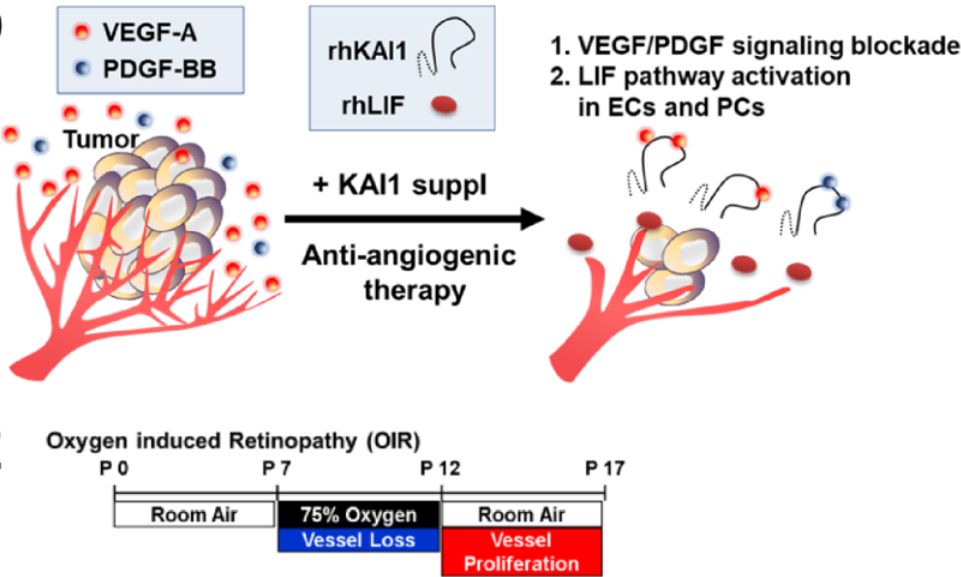
KAI1在肿瘤中的双重抗血管生成作用示意图
图片来源:https://doi.org/10.1186/s13045-021-01147-6
在本研究中,作者研究了Kai1在调控血管生成中的作用。通过对Kai1基因敲除小鼠的研究,作者证实了Kai1基因敲除促进了新生视网膜的血管生成,证明了它是抗血管生成的。通过广泛的机制研究,作者发现KAI1通过Src/P53途径在周细胞中诱导白血病抑制因子LIF。周细胞的LIF以旁分泌和自分泌的方式抑制ECs (Sox17等)和周细胞自身的一些血管生成基因。
有趣的是,Kai1还有另一种抗血管生成机制,它直接结合VEGF或PDGF并抑制它们的信号转导。在两种不同的体内肿瘤血管生成模型中,添加Kai1显著抑制肿瘤血管生成和生长,证实了Kai1的临床适用性。最后,从KAI1的大细胞外环(LEL)中提取的一种20-mer多肽已被证明具有抗血管生成的作用,在体内阻断视网膜新生血管和乳腺癌的进展。(生物谷
Bioon.com)
参考文献
Jin‑Woo Lee et al.
KAI1(CD82) is a key molecule to control angiogenesis and switch angiogenic
milieu to quiescent state. J Hematol Oncol 2021 Sep 16;14(1):148. doi:
10.1186/s13045-021-01147-6.
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
