
研究人员利用单精子长读段基因组测序进行高精度单倍型分型
来源:网络 2023-07-27 11:48
单倍型分型是指将二倍体细胞中两条同源染色体上的各种遗传多态性的连锁关系准确鉴定出来。它是终极分辨率的端粒到端粒的人类参考基因组组装的必要成分。单倍型分型能够帮助研究同一条染色体上不同调控元件遗传变异之
单倍型分型是指将二倍体细胞中两条同源染色体上的各种遗传多态性的连锁关系准确鉴定出来。它是终极分辨率的端粒到端粒的人类参考基因组组装的必要成分。单倍型分型能够帮助研究同一条染色体上不同调控元件遗传变异之间的顺式相互作用,例如,同一个基因中不同外显子上的两对杂合SNP(单核苷酸序列多态性)是位于同一条染色体上,还是分别位于两条同源染色体上,这对于判断该基因的功能状态是纯合突变状态(该基因的两个等位基因都失活)还是杂合突变状态(该基因的两个等位基因中一个失活、另外一个功能正常)非常重要。类似地,同一个基因的增强子-启动子(或者两个不同的增强子)上的两对杂合SNP的相位关系也需要通过单倍型分型来判断。
目前,最常用的单倍型分型方法是通过对指定人类个体的双亲基因组进行高通量测序实现对该个体的二倍体基因组的单倍型分型。而在缺乏亲本信息的情况下,使用长读段、超长读段或者链接读段测序可以进行基因组局部单倍型分型。然而,想要实现整条染色体尺度(长度从几千万碱基对到几亿碱基对)的单倍型分型还需要额外的技术进行辅助,例如单条染色体分选,Strand-seq和Hi-C等,这些技术可以解析出更长的遗传多态性连锁信息,提供整条染色体尺度的单倍型分型结果。然而,这些方法耗时且昂贵,相比之下,直接对单个配子细胞(单倍体细胞)进行全基因组测序是一种更加高效和精准的单倍型分型方法。
首个单精子基因组测序技术在10年前被开发出来,此后单精子基因组测序技术被不断优化和改进,为减数分裂、生殖细胞基因组不稳定性和单倍型分型的研究提供了强有力的技术支持。然而目前为止几乎所有的单精子基因组测序方法都仅限于二代测序平台,由于所获得的序列长度不超过600bp,对于单倍型分型的研究只限于SNP水平,对于基因组不稳定性的研究几乎都集中在基因组拷贝数变异(CNV)和单碱基突变上(SNV),对于基因组上更重要的遗传多态性(例如结构变异,特别是50bp—10kb的结构变异)的鉴定和分型很难实现。
2023年6月24日,北京大学生物医学前沿创新中心(BIOPIC)汤富酬教授课题组在Nucleic Acids Research发表了题为“Long-read-based single sperm genome sequencing for chromosome-wide haplotype phasing of both SNPs and SVs”的研究论文。该研究开发了一种基于单分子测序(三代测序)平台的单精子长读段基因组测序方法和相应的数据分析流程,通过高质量的单精子基因组测序数据能够准确鉴定精子减数分裂过程中的交叉重组事件,并且能够准确检测单个精子细胞基因组中的结构变异。更重要的是,单精子长读段基因组测序实现了整条染色体尺度的单倍型分型(包括SNP和结构变异)(图1)。

文章封面

图1. 基于长读段测序平台的单精子基因组测序方法及应用
1. 首次基于单分子测序平台开发了一种高通量单精子长读段基因组测序方法(同时适用于ONT和PacBio单分子测序平台)。该研究设计了24种带有不同条码序列的Tn5酶,并结合后续扩增引物中使用的96种条码序列,对单个精子进行双重条码序列标记,使得在一次测序中通量最高可以达到2304个单细胞。然后将24个带有不同Tn5酶转座标记的精子混合在一起进行基因组扩增,以增加扩增的均匀性,并进行了严格的交叉污染实验,验证了该方法的低交叉污染率和可靠性。最终从B6D2F1/Crl [BDF1]雄性杂合小鼠中获取了共1573个精子细胞的基因组测序数据(过滤掉含有多个精子细胞或基因组覆盖度低于1%的样本)。在平均测序深度只有0.1×的情况下,单个精子的基因组覆盖度为1%到25.5%(基因组覆盖度中位数为4.9%),平均测序读长为5.5kb(图2)。
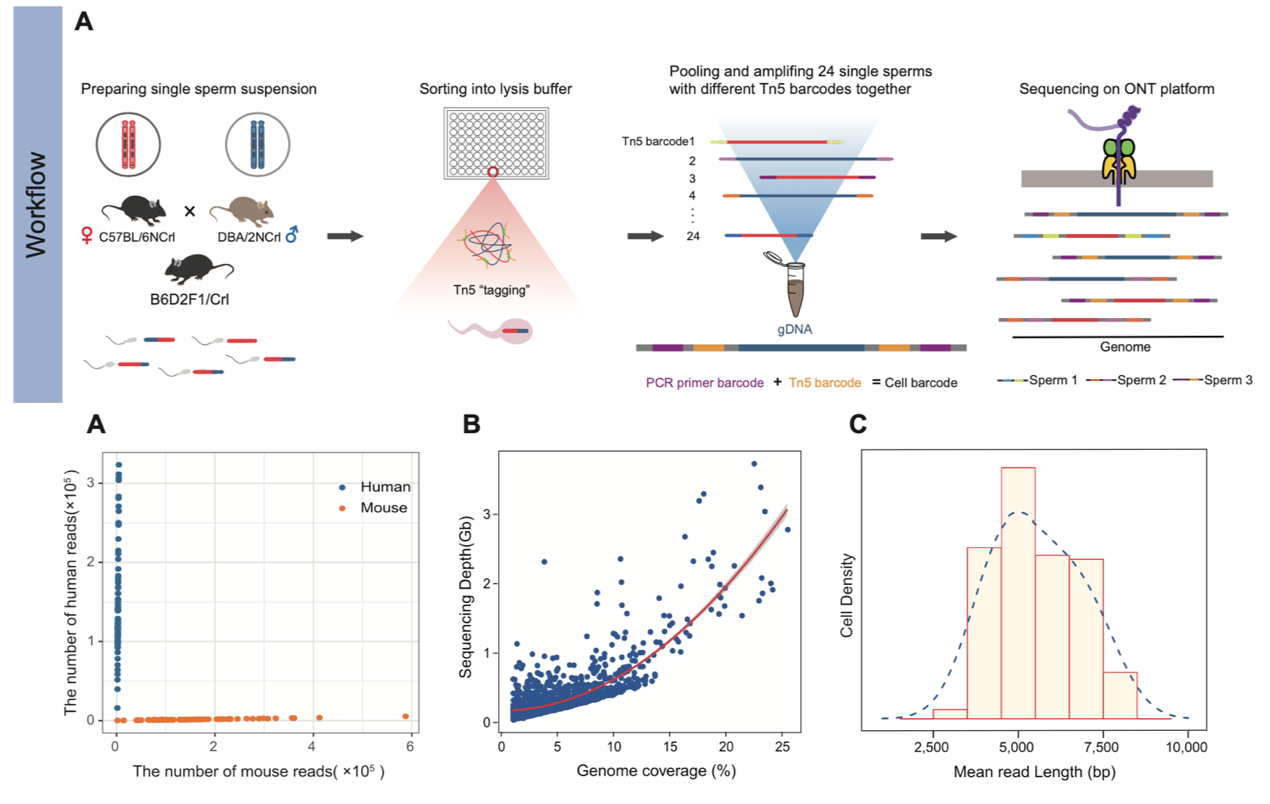
图2. 基于长读测序平台的单精子基因组测序方法实验流程图和数据质控
2. 精准鉴定出每个精子细胞中的减数分裂交叉重组事件和非整倍体事件。该研究使用隐马尔可夫模型(HMM)来确定单个精子中出现的减数分裂交叉重组位点。最终在1573个精子样本中鉴定出17,445个常染色体交叉重组事件。每个精子会发生4到27次交叉重组(平均每个精子发生12次)。同时在单精子基因组的测序数据中,也鉴定出了交叉干涉现象。同时,在23个精子细胞中鉴定到29个整条染色体丢失事件,其中15个发生在常染色体上,14个发生在性染色体上,此外,还在4个精子细胞中发现了4个常染色体拷贝数增加事件(图3)。
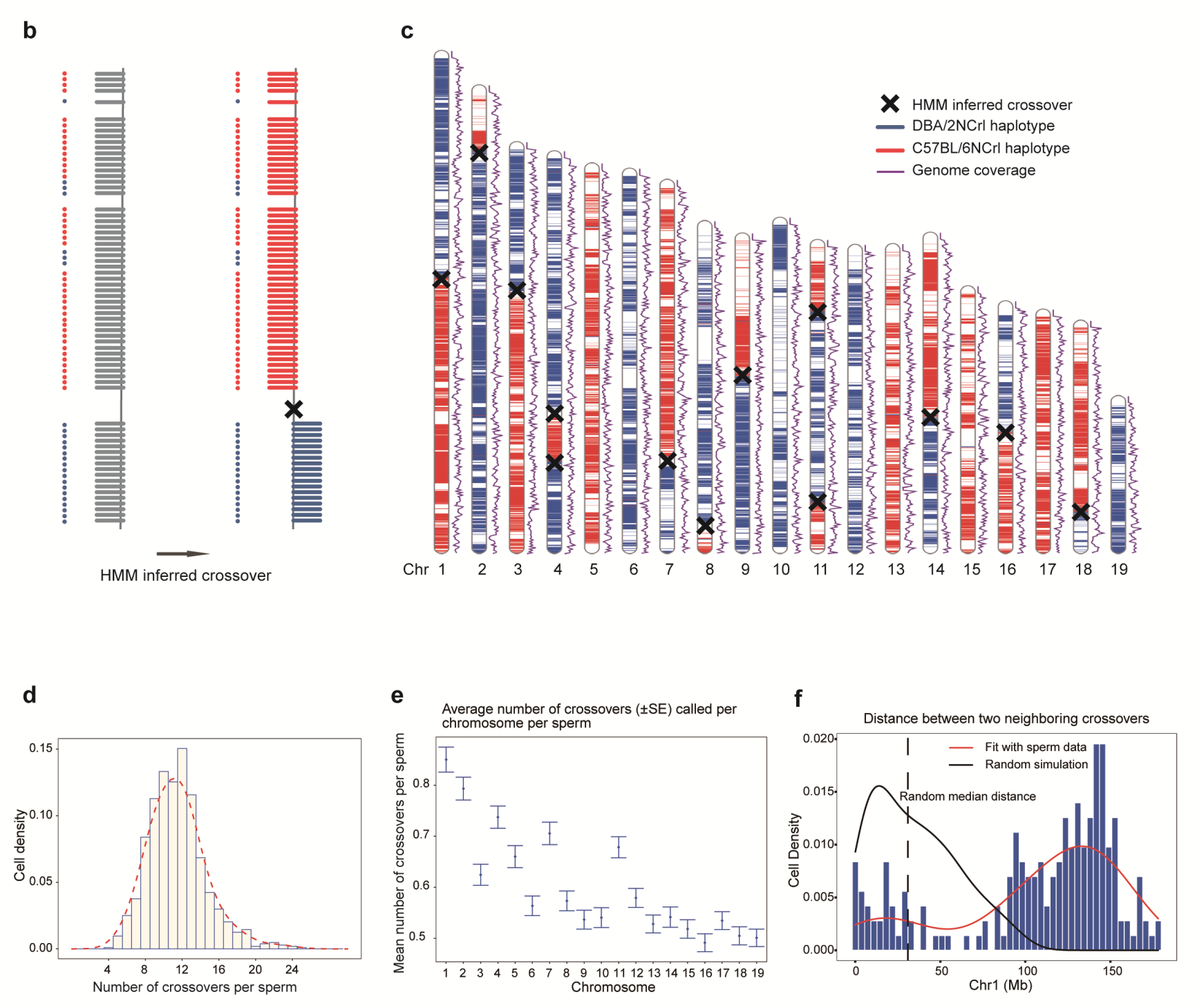
图3. 单精子中交叉重组事件的鉴定
3. 在单个精子细胞中精确鉴定出结构变异(SV)。该研究以父母本小鼠大量细胞起始(bulk)的ONT单分子测序数据所鉴定出的结构变异作为金标准(DBA/2NCrl小鼠和C57BL/6NCrl小鼠基因组中分别鉴定出57,116个和5901个结构变异),来评估单精子基因组测序数据中结构变异检测的准确性,发现6个以上精子细胞支持的结构变异事件的检测精度可以到达90%,而3个精子细胞支持的结构变异F1评分最高(78%)。另外,该研究选取了70个涉及重复元件的结构变异进行PCR实验验证,验证准确性达到90%左右(图4)。
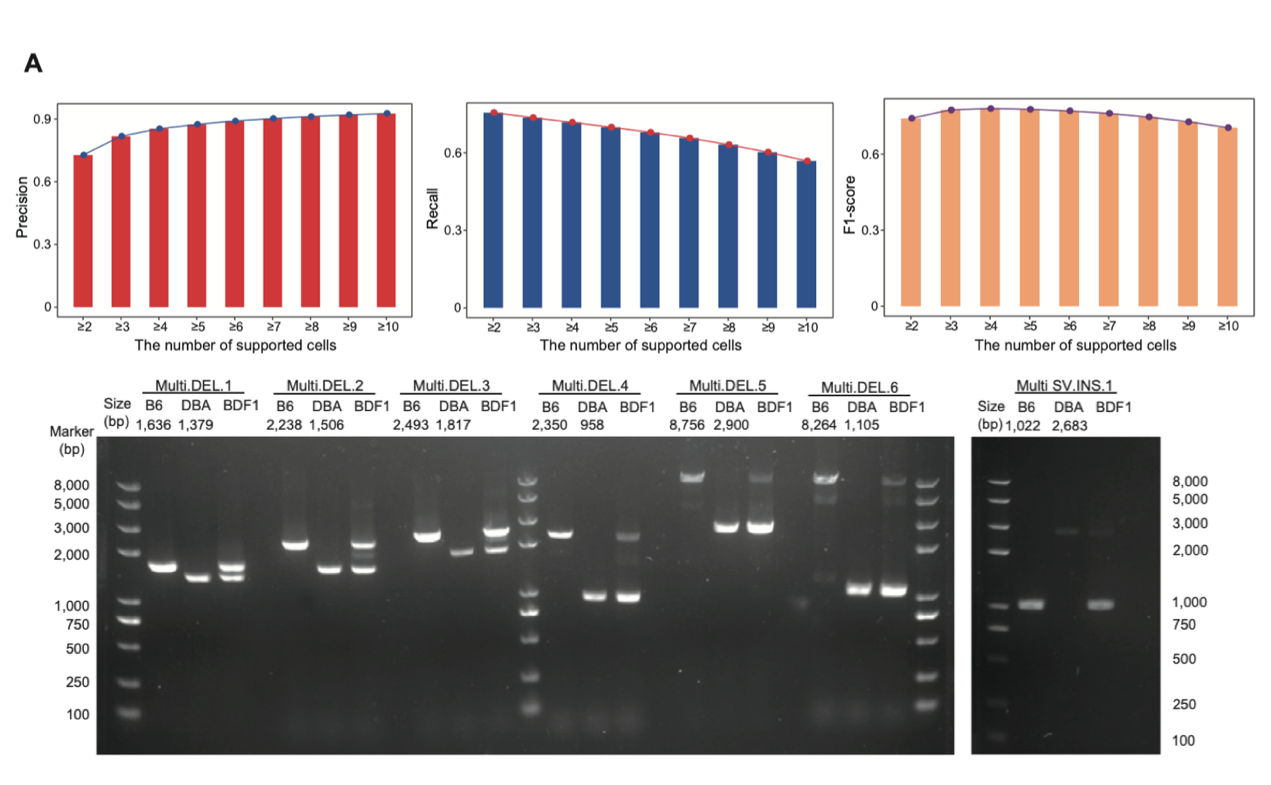
图4. 单精子中基因组结构变异的鉴定
4. 实现了对SNP和结构变异等遗传多态性的整条染色体尺度的高精度单倍型分型。该研究开发了一种新的染色体尺度的单倍型分型流程。在该流程中,总共鉴定出54,712个杂合结构变异,其中94.36%的杂合结构变异能够通过该技术进行分型,分型准确率达到98.59%。总共鉴定出4,664,507个杂合SNP,其中98.15%的杂合SNP能够通过该技术进行分型,分型准确率达到99.95%。同时,成功进行分型的结构变异的长度分布显示出一个约190bp的峰值和一个6kb的峰值,它们分别对应了哺乳动物基因组中两种数量较多的重复元件:SINE和LINE(图5)。
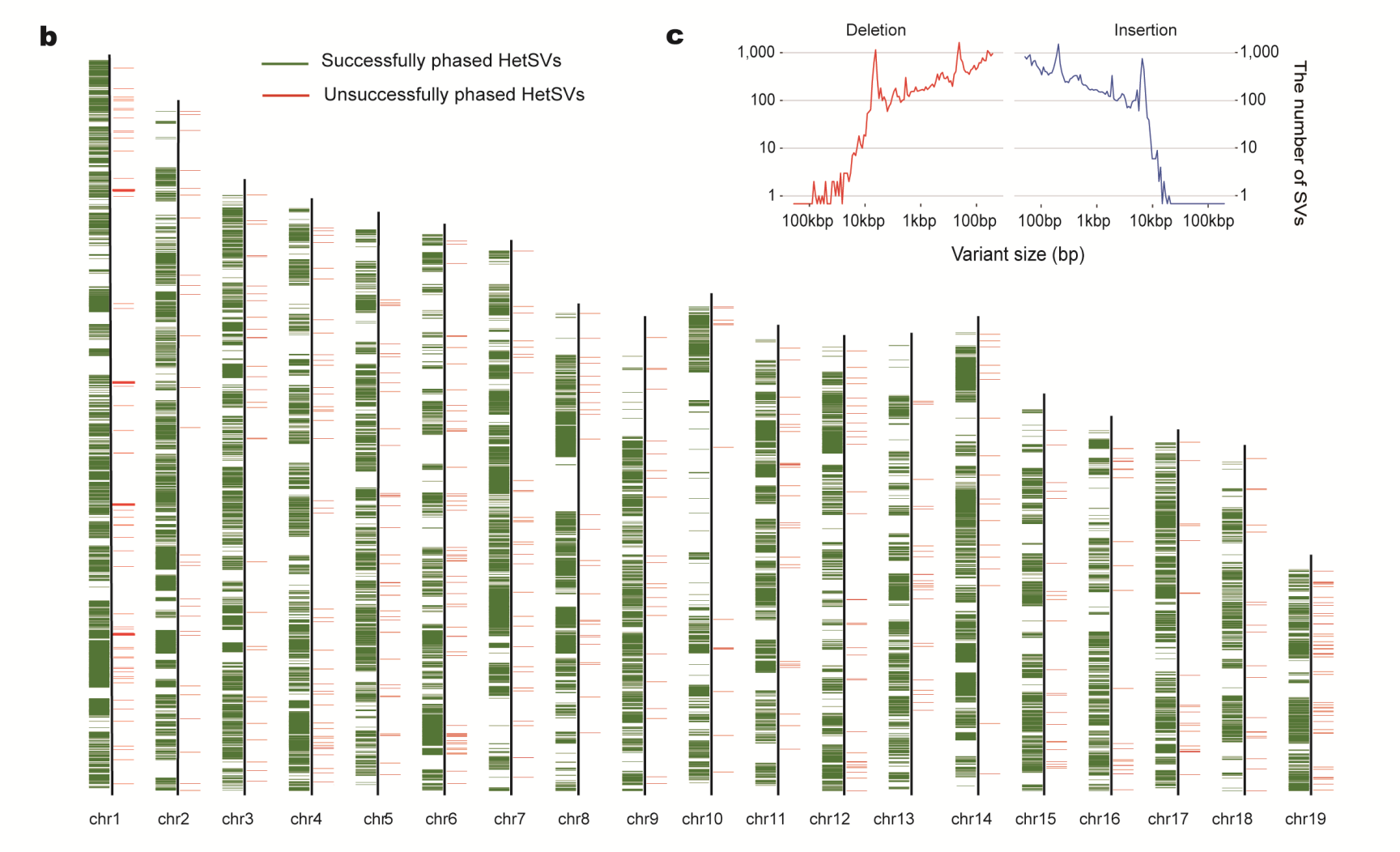
图5. 使用单精子基因组测序数据完成染色体尺度的单倍型分型
5. 精准识别和分类包含重复元件的结构变异。在长度大于100bp的36,271个成功进行分型的杂合结构变异中,25,664个包含重复元件。其中,29.3%被单个LINE覆盖,17.0%被单个SINE覆盖,16.2%被单个LTR覆盖,12.4%被串联重复覆盖,24.9%被多个重复元件的组合覆盖。同时鉴定出3190个串联重复序列在单倍型之间存在特异性扩增,为此该研究使用k-mer频率信息来可视化这些串联重复位点在B6和DBA单倍型中的结构和序列组成差异(图6)。
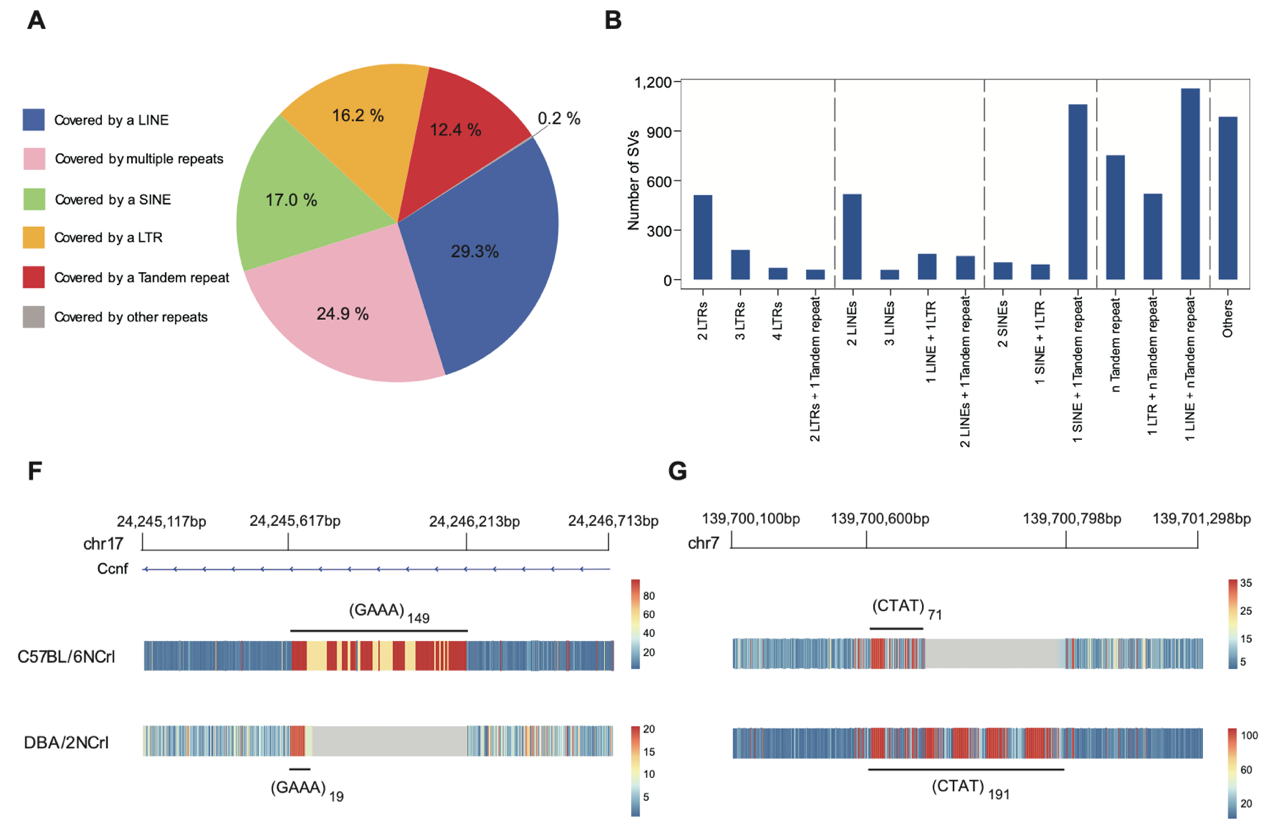
图6. 涉及重复元件的基因组结构变异鉴定和可视化
该研究有几个潜在的应用:首先,类似于Strand-seq技术,该方法可以与大量细胞起始(bulk)的长读段基因组测序相结合,实现高精度的单倍型组装。或者通过对数百个单个精子细胞进行高深度长读段基因组测序,直接完成单倍型从头组装。其次,该方法可以应用于人类单个精子细胞的研究,通过在不育男性生殖细胞中鉴定基因组结构变异(包含重复元件),可以为男性不育的研究和遗传疾病的研究提供新的线索。第三,该研究结果显示,最少只需要100个精子细胞就能完成整条染色体尺度的高精度单倍型分型,这大大降低了单倍型分型的成本,为未来构建人类泛基因组图谱开辟了新的途径。
北京大学生物医学前沿创新中心、北京大学生命科学学院博士谢昊伶,北京大学前沿交叉学科研究院博士生李文以及北京大学生命科学学院博士生郭雨晴为该论文的并列第一作者。汤富酬为该论文的通讯作者。该研究项目得到了北京大学生物医学前沿创新中心、北京未来基因诊断高精尖创新中心、北大-清华生命科学联合中心、北京昌平实验室、北京市科技委的支持。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
