
乳酸菌竟是心梗救治的隐形卫士?BMC Med研究揭秘:通过多重机制减轻心肌缺血再灌注损伤
来源:生物谷原创 2025-07-11 11:36
乳酸菌可激活相关通路,多靶点抑制心肌缺血再灌注损伤,为心梗治疗提供新策略 。
急性心肌梗死(AMI)作为威胁全球健康的高危心血管疾病,其高死亡率和并发症发生率始终是临床治疗的难题。目前,早期冠状动脉血运重建以恢复缺血心肌血流是主要治疗手段,但血流恢复过程中引发的心肌缺血再灌注(I/R)损伤,可导致高达50%的心肌梗死面积扩大,成为改善患者预后的重大障碍。
近期,发表于BMC Med的一项研究Lactobacillus ameliorates myocardial ischemia reperfusion injury by attenuating apoptosis, inflammation, oxidative stress, and ferroptosis揭示:乳酸菌或能通过多重机制减轻心肌I/R损伤,为临床治疗提供了新方向。

该研究结合临床观察、动物实验和细胞实验,系统探究了乳酸菌对心肌I/R损伤的保护作用及机制。在纳入158例ST段抬高型心肌梗死(STEMI)患者的临床研究中,发现乳酸菌水平与心肌损伤标志物(肌酸激酶、肌酸激酶同工酶MB、乳酸脱氢酶、B型脑钠肽)、炎症因子(肿瘤坏死因子-α、白细胞介素-6、白细胞介素-1β)及丙二醛呈显著负相关,而与谷胱甘肽呈正相关。这表明体内乳酸菌含量越高,心肌损伤、炎症反应及氧化应激程度越轻。
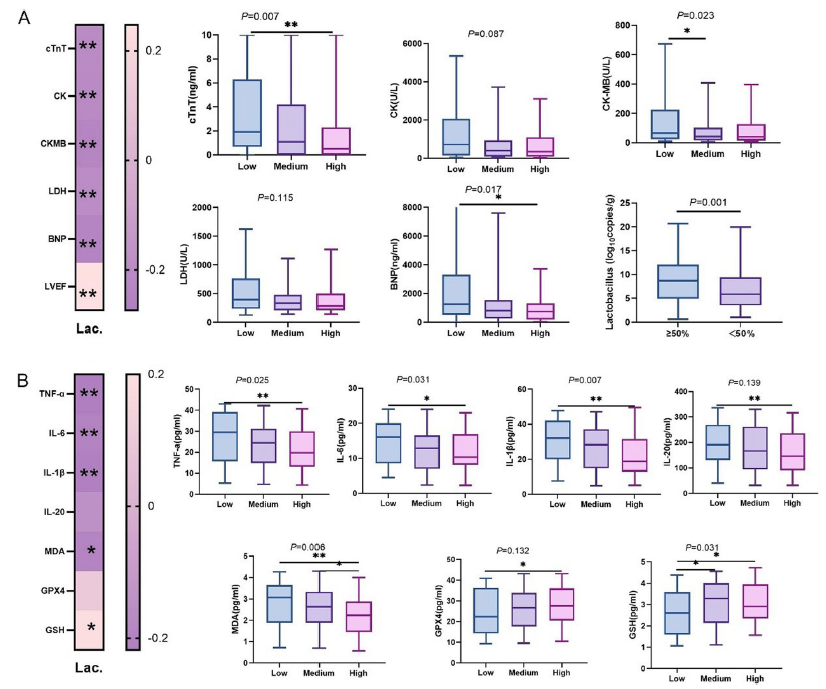
图 1:乳酸菌与心肌损伤标志物及血清学因子的关系
为验证乳酸菌的直接作用,研究团队对大鼠进行了4周乳酸菌干预后构建心肌I/R模型。结果显示,乳酸菌干预显著降低了大鼠心肌损伤标志物水平,缩小梗死面积,改善心肌细胞排列紊乱,并提升心功能(左心室射血分数、缩短分数显著提高,左心室舒张末期和收缩末期内径减小)。进一步机制研究发现,乳酸菌通过上调抗凋亡蛋白Bcl-2、下调促凋亡蛋白Bax和半胱天冬酶-3,抑制心肌细胞凋亡;同时降低炎症因子水平,减少丙二醛和活性氧生成,提高超氧化物歧化酶活性,缓解氧化应激。
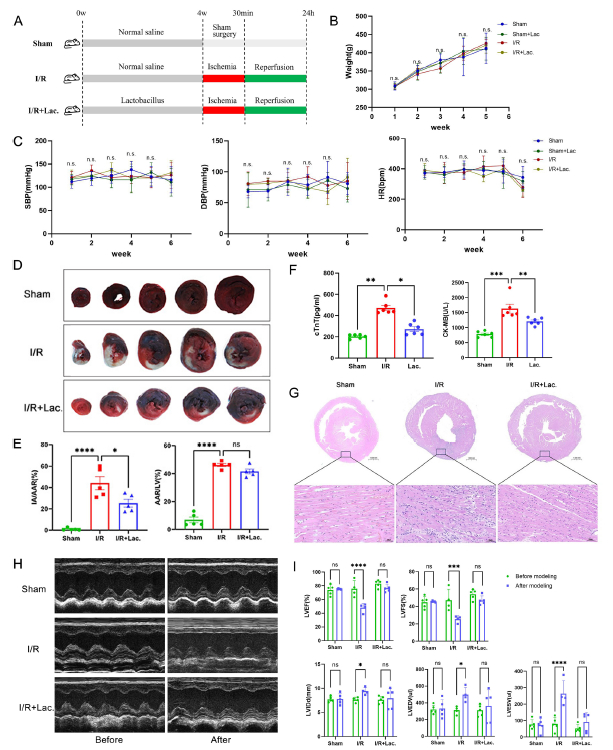
图 2:乳酸菌改善心肌缺血再灌注损伤
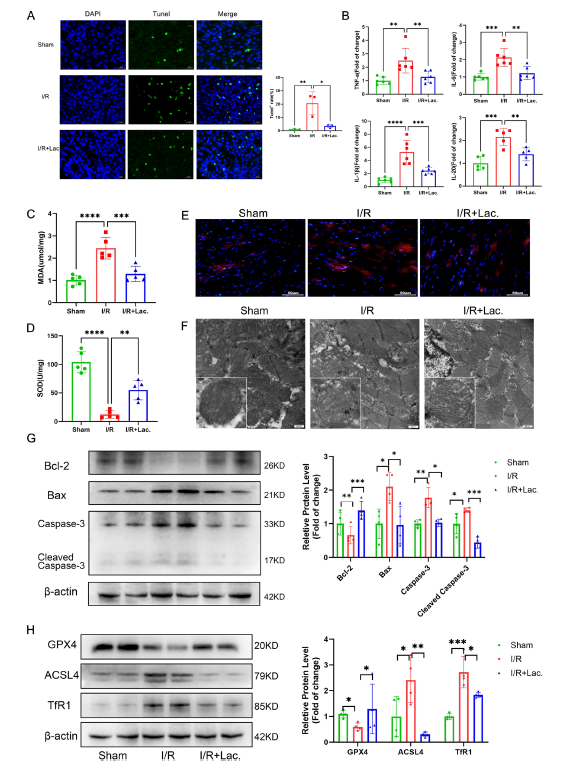
图 3:在大鼠心肌缺血再灌注模型中,乳酸菌通过抑制凋亡、炎症、氧化应激和铁死亡改善心肌缺血再灌注损伤
值得注意的是,该研究首次明确乳酸菌对ferroptosis(铁死亡)的抑制作用。铁死亡是一种铁依赖的脂质过氧化累积导致的程序性细胞死亡,与心肌I/R损伤密切相关。实验中,乳酸菌可上调谷胱甘肽过氧化物酶4(GPX4)的表达,同时下调长链脂酰辅酶A合成酶4(ACSL4)和转铁蛋白受体1(TfR1),从而减少脂质过氧化和铁蓄积,减轻铁死亡引发的心肌损伤。
为揭示深层分子机制,研究通过敲除和过表达实验,证实乳酸菌的保护作用依赖于Sirtuin 1(Sirt1)/核因子erythroid 2相关因子2(Nrf2)/血红素氧合酶1(HO-1)信号通路。在心肌细胞中,乳酸菌可激活Sirt1,促进Nrf2去乙酰化并进入细胞核,进而上调HO-1的表达。这一通路的激活不仅增强了细胞的抗氧化和抗炎能力,还协同抑制了凋亡和铁死亡过程。当Sirt1或Nrf2被敲除时,乳酸菌的保护作用显著减弱;而过表达Sirt1或Nrf2则可模拟乳酸菌的有益效果,进一步验证了该通路的核心地位。
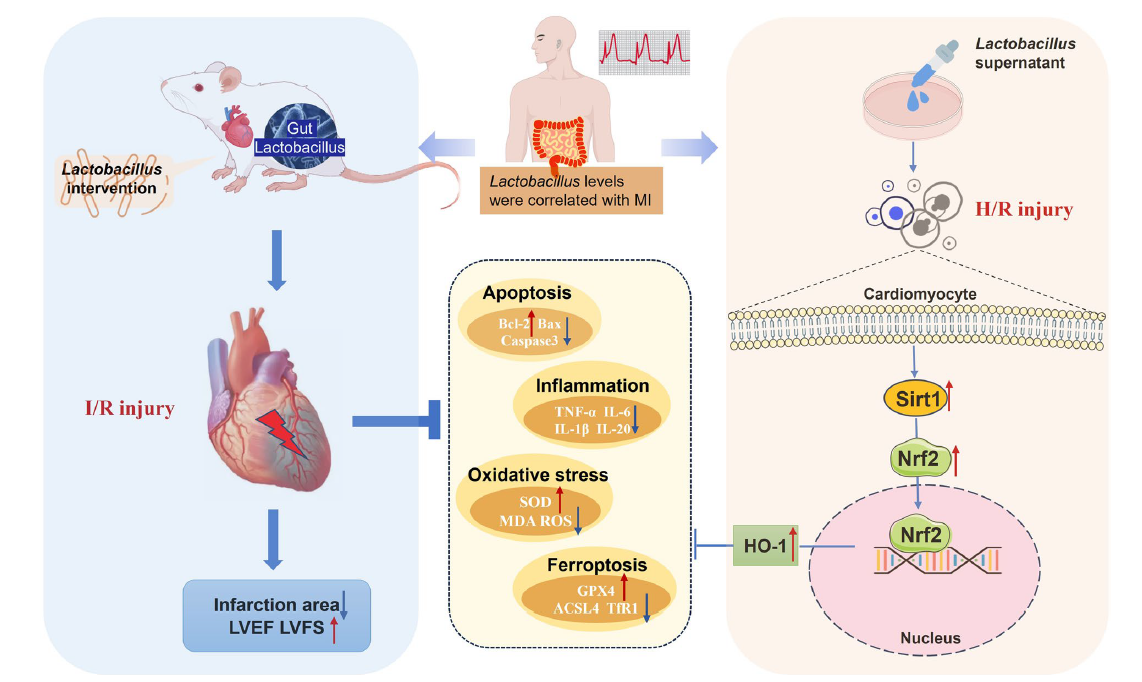
图 4:乳酸菌对心肌缺血再灌注损伤保护作用的示意图
综上,这项研究从临床到基础全面证实,乳酸菌通过激活Sirt1/Nrf2/HO-1通路,多靶点抑制心肌I/R损伤中的凋亡、炎症、氧化应激和铁死亡,从而发挥心脏保护作用。对于心血管疾病患者而言,调节肠道菌群中乳酸菌的丰度或许成为改善心肌再灌注损伤的新策略。未来,随着对乳酸菌代谢产物及临床应用方案的深入研究,有望为急性心肌梗死患者的治疗提供更精准、有效的干预手段,助力降低心血管事件的致残率和死亡率。(生物谷Bioon.com)
参考文献:
Liang Y, Zhao L, Zhang X, et al. Lactobacillus ameliorates myocardial ischemia reperfusion injury by attenuating apoptosis, inflammation, oxidative stress, and ferroptosis. BMC Med. 2025;23(1):377. Published 2025 Jul 1. doi:10.1186/s12916-025-04203-x
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
