
《自然·免疫学》:刷新认知!IFN-γ信号竟然会抑制杀伤性T细胞抗肿瘤活性!
来源:奇点糕 2022-11-22 13:20
这项研究发现了IFN感应性的丧失在多个动物模型中使得肿瘤对ICB更加敏感。这可以通过两种机制介导,第一是经典MHC-I类分子的上调能够抑制NK细胞,第二是非经典MHC-I类分子Qa-1b的上调可以通过
免疫检查点阻断(ICB)疗法在多种肿瘤中都取得了巨大成功。然而,仍有许多患者对ICB没有响应,不同肿瘤中的免疫逃逸机制也需要进一步探索。
近日,由布罗德研究所(Broad Institute)的Robert T. Manguso和Kathleen B. Yates带领的研究团队,在著名期刊Nature Immunology发表重要研究成果[1]。
为了发现肿瘤逃避免疫治疗的机制,他们在接受ICB治疗的癌症模型中,开展了基因组层面的CRISPR筛选。
他们的发现让他们大吃一惊:在多种肿瘤模型中,肿瘤细胞对干扰素(IFN)感应性的丧失,竟然使得肿瘤对ICB的响应更加敏感。
具体来说,肿瘤的IFN信号通过两种方式发挥免疫抑制的作用:第一,肿瘤IFN感应能够上调经典的第一类主要组织相容性复合体(MHC-I)抑制自然杀伤(NK)细胞;第二,肿瘤IFN感应通过上调非经典的MHC-I类分子Qa-1b抑制CD8+T细胞。
要知道,大量的研究表明IFN-γ在肿瘤的免疫监视中发挥重要作用[2-4]。这一发现可以说颠覆了我们对IFN-γ信号的认知。

论文首页截图
接下来我们就一起来看看这个研究是如何开展的。
研究团队利用代表了5种癌症类型(黑色素瘤、胰腺癌、肺癌、肾癌和结肠癌)的8个小鼠肿瘤模型,进行CRISPR体内筛选,找出ICB治疗后富集或清除的sgRNA,以鉴定与ICB抵抗性或敏感性相关的基因(指该基因缺失时分别与ICB抵抗性或敏感性相关)。
他们发现Nlrc5、Pten、Casp8、Ccar1、Ubr5、Dot1lh、Smg9和Pdcd10的缺失与ICB抵抗性相关,并对Ccar1缺失这一新发现的ICB抵抗机制进行体内实验验证。
在ICB敏感性方面,除了已报道的Ptpn2、Traf2、Ripk1、Serpinb9、Cd47、Atg7外,研究团队还发现了Calr、Rnf31、Vps45、Ilf2、Pnpo、Kcmf1、Fitm2、Morc2a和Thop1的缺失与ICB敏感性相关,并进一步对Calr、Med16和Rnf31缺失介导的ICB敏感机制进行体内实验验证。
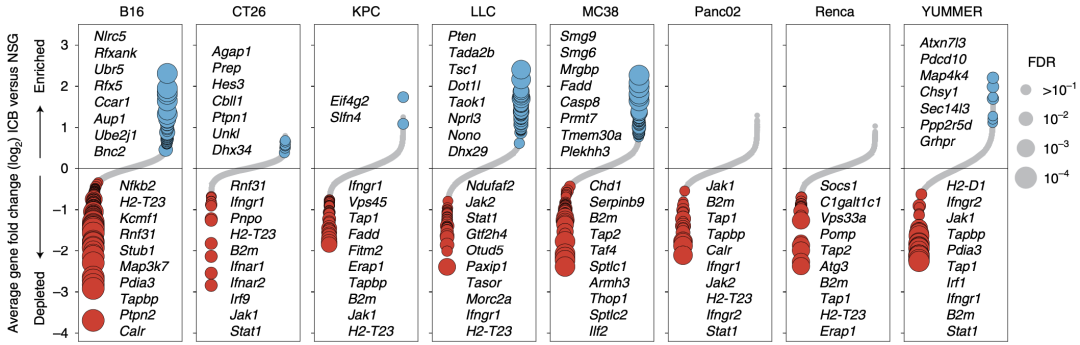
CRISPR筛选发现的与ICB抵抗性或敏感性相关的基因
研究团队进一步对清除的sgRNA进行了通路富集分析,令人意外的是,MHC-I类抗原呈递信号通路相关的基因在除Lewis肺癌细胞(LLC)外的所有模型中都被清除,细胞因子响应、II型IFN和先天免疫相关的基因也在多个模型中被清除。这提示这些基因的缺失可以使得肿瘤细胞对ICB更加敏感。
我们都知道,IFN-γ可以上调MHC-I类分子的表达,从而促进细胞毒性CD8+T细胞(CTL)识别肿瘤细胞[4]。还有很多研究表明,IFN感应性的丧失与ICB的抗性相关[2,3,5,6]。在类似的小鼠肿瘤模型中进行体外CRISPR筛选,发现肿瘤细胞IFN和抗原呈递通路的丧失会引起肿瘤细胞对CTL杀伤的抵抗[7]。而本研究发现抗原呈递和IFN通路相关基因的清除竟然与ICB的敏感性相关,这似乎与上述研究结果相矛盾。
为了搞清楚矛盾背后的原因,Manguso团队进一步对比了他们的体内CRISPR筛选结果和前人的体外CRISPR筛选结果,结果发现了一些与IFN(如Jak1、Ifngr1、Ifngr2)和抗原呈递(如Tap1、Tap2、B2m)相关的基因,在体外和体内CRISPR模型中分别发生了富集和清除。
这一发现提示,在体外研究中,这些基因的缺失可能促进了肿瘤细胞对T细胞杀伤的抵抗;而在体内研究中,这些基因的缺失让肿瘤细胞对T细胞更敏感。
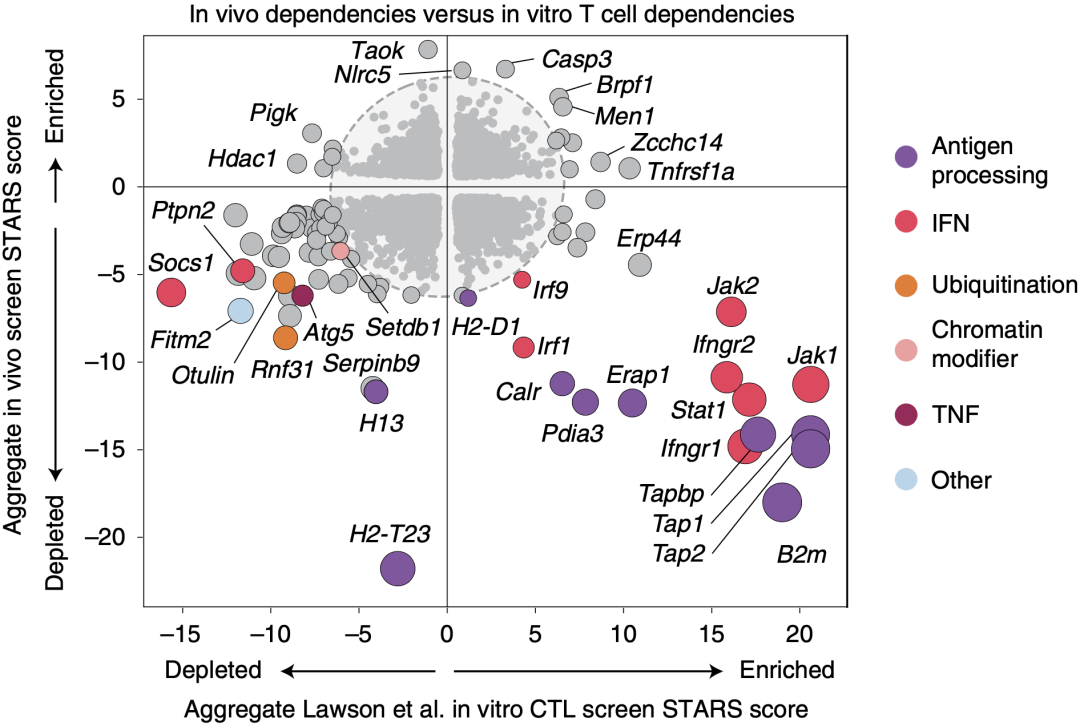
体外模型与体内模型中CRISPR筛选得到的sgRNA对比
为了进一步探究IFN诱导肿瘤细胞对ICB抵抗的机制,Manguso团队对比了ICB治疗后的清除的sgRNA与IFN-γ刺激后改变的IFN刺激基因(ISG)。结果显示,抗原呈递相关基因(H2-T23、B2m、Tap1、Tap2、Tapbp和Erap1)在ISG中富集,这些基因同样在ICB治疗后清除的sgRNA中富集,这提示这些基因能够被IFN-γ刺激诱导上调,并与ICB治疗的抵抗性相关。
体外实验也说明,在IFN-γ或IFN-β刺激后,经典MHC-I类分子H-2K、H-2D,非经典MHC-I类分子Qa-1b以及PD-L1的表达会发生上调。这些结果提示IFN诱导的ICB抵抗机制可能是由MHC-I类分子的上调所介导的。
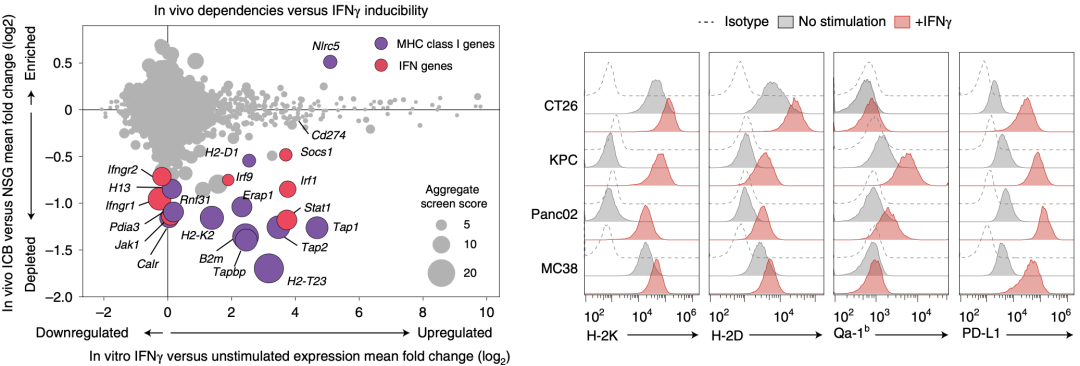
IFN-γ刺激能够诱导多种发挥ICB抵抗性的基因上调
为了验证这一假设,研究团队设计了一种体内竞争性实验,以检测IFN-γ感应基因和MHC-I类抗原呈递途径相关基因的遗传上位性。
他们将对照sgRNA和靶向Ifngr1或Jak1(IFN-γ感应基因)的sgRNA转染的KPC细胞(这些细胞分别是对照sgRNA背景、Tap1敲除背景和B2m敲除背景,Tap1和B2m为MHC-I类抗原呈递途径相关基因)1:1混合,然后将这些细胞分别在NSG鼠,WT鼠和接受ICB治疗的WT鼠体内成瘤,对比不同组中转染对应sgRNA的细胞的富集与清除。
结果显示,在对照sgRNA背景的细胞中,Ifngr1或Jak1敲除的细胞被清除;而在Tap1和B2m敲除背景的细胞中,Ifngr1或Jak1敲除的细胞没有发生显著清除。这提示相比于对照细胞,IFN感应性缺陷的细胞具有依赖于MHC-I类的竞争劣势,即IFN感应性通路发挥功能依赖于MHC-I类分子的表达。
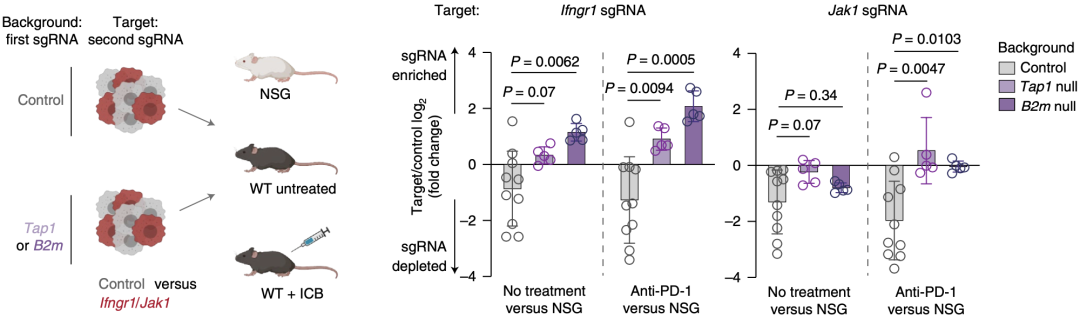
IFN-γ感应基因发挥功能依赖于MHC-I类抗原呈递途径相关基因
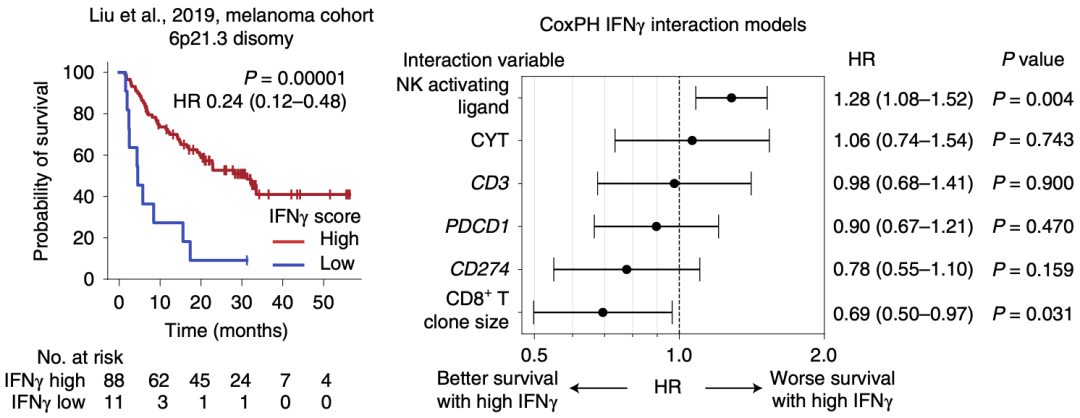
在人类基因组中,6p21.3基因座编码MHC-I类分子表达相关的基因,如TAP1、TAP2、TAPBP、PSMB8和PSMB9等[8-10]。TCGA中多种癌种都有超过10%的肿瘤发生了6p21.3缺失,然而6p21.3缺失与绝大多数肿瘤患者的生存无关。
在接受抗PD-1治疗的肾透明细胞癌患者队列中,当肿瘤为6p21.3两倍性时,高ISG特征与患者更差的生存相关。然而在6p21.3缺失的肿瘤中,高ISG特征不再与患者更差的生存相关。这提示肾透明细胞癌中,IFN介导的ICB抵抗作用依赖MHC-I类分子。
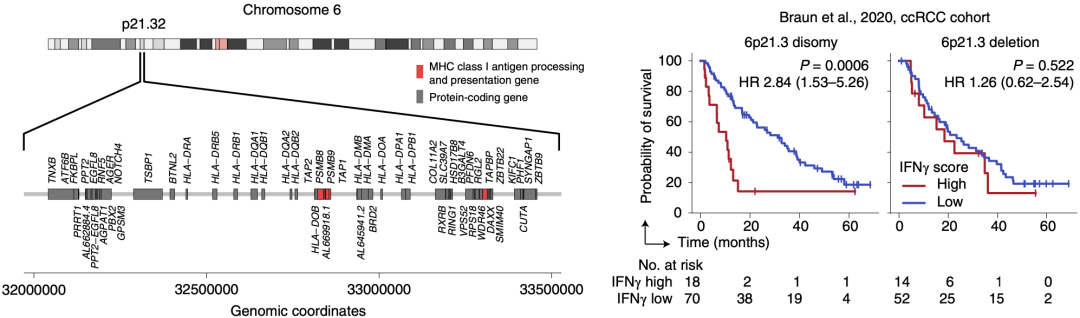
肾透明细胞癌中高ISG特征仅在6p21.3两倍性肿瘤中与患者差的预后相关
研究团队为进一步鉴定体内杀伤IFN感应性缺陷肿瘤的免疫细胞,分别在ICB治疗时使用抗体清除CD8+T细胞、NK细胞和CD4+T细胞,结果显示NK细胞和CD4+T细胞的清除影响了对Jak1缺陷肿瘤细胞的杀伤,而清除CD8+T细胞对Jak1缺陷肿瘤细胞的杀伤没有影响。由于NK细胞不表达PD-1,而CD4+T细胞表达PD-1,研究团队推测ICB治疗后,CD4+T细胞辅助NK细胞发挥肿瘤细胞杀伤作用。
为进一步在体外验证NK细胞对IFN感应性缺陷肿瘤的杀伤能力,他们将Jak1缺陷和对照肿瘤细胞,分别与IL-12/18激活的NK细胞共培养,结果显示活化的NK细胞能够杀伤Jak1缺陷肿瘤细胞,而肿瘤细胞中Tap1或H2-K1的缺失可以逆转这一效应。这提示肿瘤IFN感应性可以通过上调经典的MHC-I类分子,在体内和体外抑制NK细胞的毒性。
Manguso团队进一步探究了哪些MHC-I类抗原呈递途径的基因发挥功能依赖于NK细胞,他们对比了NK细胞清除与不清除时,ICB治疗清除的sgRNA。结果显示,尽管Ifngr1、Ifngr2、Stat1、Jak1、Jak2和H2-K1的清除表现出NK细胞依赖性,靶向Tap1、Tap2、H2-D1和H2-T23这几个抗原呈递基因的sgRNA在NK细胞存在与不存在时都会发生清除。这说明部分MHC-I类抗原呈递分子的缺失,所引起的肿瘤细胞杀伤是非NK细胞依赖性的。
在上述基因中,H2-T23引起了Manguso团队的关注。因为H2-T23编码非经典MHC-I类分子Qa-1b(相当于人类中的人类白细胞抗原HLA-E),而Qa-1b能够结合抑制性受体NKG2A/CD94。这些结果提示尽管NK细胞介导了对IFN感应性丧失的细胞的杀伤,但是某些MHC-I类分子(如Qa-1b)的表达可能会抑制其他的效应性细胞群。
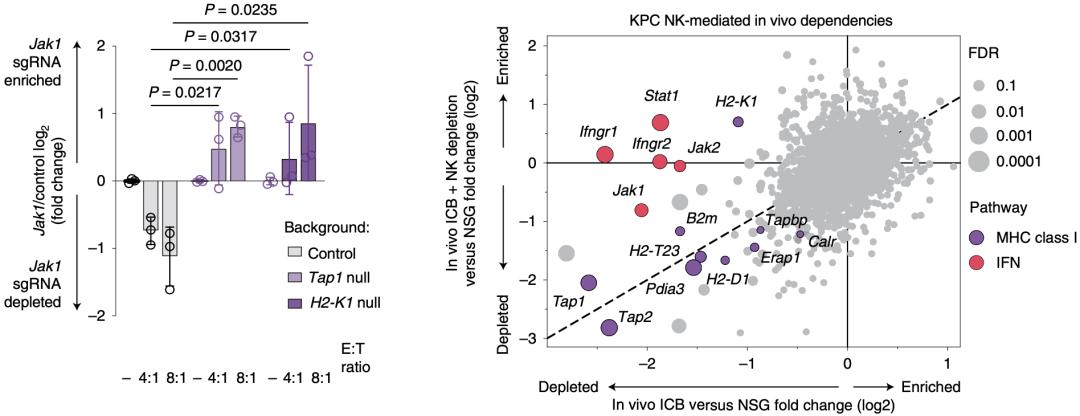
某些MHC-I类分子(如Qa-1b)的表达可能会抑制除NK细胞外的效应性细胞
为了探究Qa-1b表达抑制了哪些效应性细胞群,研究团队首先检测了Qa-1b的缺失是否会增强ICB响应。结果显示,Qa-1b的缺失会增强ICB响应,而Qa-1b的过表达能抑制ICB响应。
当在接受ICB治疗的小鼠胰腺导管腺癌(KPC)肿瘤模型中敲除HT-T23后,分别清除NK细胞和CD8+T细胞,结果显示NK细胞清除对于Qa-1b缺陷型肿瘤的ICB响应没有影响,而CD8+T细胞清除抑制了Qa-1b缺陷型肿瘤的ICB响应。这提示Qa-1b主要通过抑制CD8+T细胞发挥作用。
单细胞测序数据也提示,ICB治疗后引起效应、耗竭和增殖的CD8+T细胞亚群扩增。而效应、耗竭和增殖CD8+T细胞亚群高表达NKG2A(Klrc1)和CD94(Klrd1),这提示肿瘤细胞的Qa-1b表达可能通过与CD8+T细胞的NKG2A/CD94抑制性受体结合发挥作用。
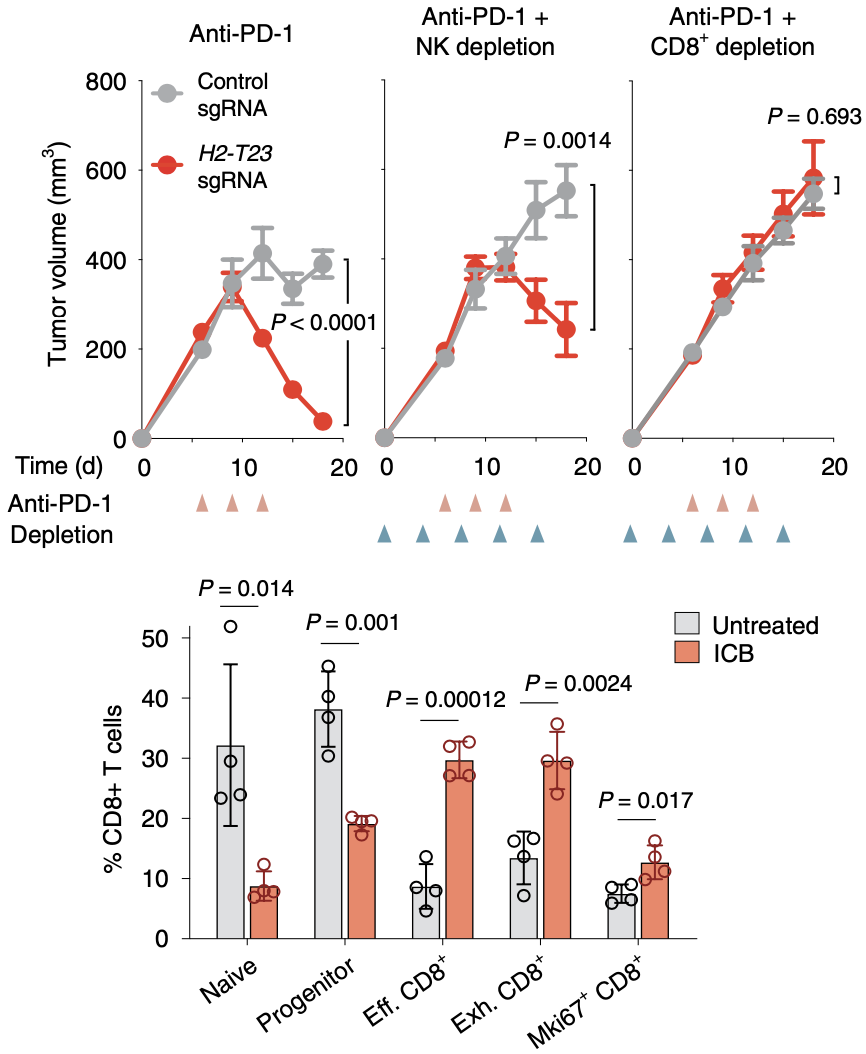
ICB治疗时CD8+T细胞发挥杀伤Qa-1b缺陷肿瘤作用
由于IFN能够诱导Qa-1b的表达,Manguso团队假设肿瘤的IFN感应性可以通过Qa-1b抑制CD8+T细胞。然而由于肿瘤IFN感应性丧失可能引起经典的MHC-I类分子下调,导致对CD8+T细胞的抗性,从而掩盖IFN可能介导的抑制作用。为解决这个问题,利用嵌合抗原受体(CAR)T细胞进行杀伤试验,由于CAR-T细胞的识别不依赖于MHC-I类分子,可以避免IFN-γ处理后MHC-I类分子上调带来的影响。
他们首先检测了CD19 CAR-T细胞对Qa-1b缺陷型CD19+KPC细胞的杀伤能力,结果显示CD19 CAR-T显著杀伤Qa-1b缺陷型肿瘤细胞,提示Qa-1b能够抑制CAR-T的杀伤能力。
为探讨Qa-1b上调是否是由IFN介导的CAR-T细胞杀伤抵抗机制,Manguso团队将CD19 CAR-T细胞分别与IFNγ刺激的对照和Jak1(IFN-γ感应基因)敲除CD19+KPC细胞的混合物1:1共培养。结果显示,CAR-T细胞对于这2类细胞的杀伤能力没有显著差异,这表示CAR-T细胞消除了Jak1敲除肿瘤细胞的生存优势。
为了探究IFN诱导的PD-L1上调是否抑制CAR-T的杀伤能力,他们检测了CAR-T联合抗PD-1单抗时两组细胞的存活情况。结果显示当CAR-T联合抗PD-1单抗时,Jak1敲除肿瘤细胞存活增多,这提示PD-L1上调对T细胞杀伤能力发挥抑制作用,联用抗PD-1单抗能够杀伤更多对照组肿瘤。
当Manguso团队使用Qa-1b缺陷型KPC细胞或联用抗NKG2A抗体时,结果显示Jak1敲除肿瘤细胞存活显著增多。这些结果提示CD8+T细胞优先杀死具有完整IFN感应能力的肿瘤细胞,而Qa-1b的上调抑制这种杀伤能力。至于背后的机制,Manguso团队认为这可能是因为IFN能够诱导粘附分子(如ICAM-1)的表达[11],进而促进CD8+T细胞的杀伤。
在体内实验模型中,Manguso团队发现CAR-T对Jak1缺陷型肿瘤杀伤作用明显,而这种杀伤能力在Qa-1b缺陷的肿瘤中消失。这些结果提示IFN信号可以通过上调Qa-1b来抑制CAR-T细胞毒性。
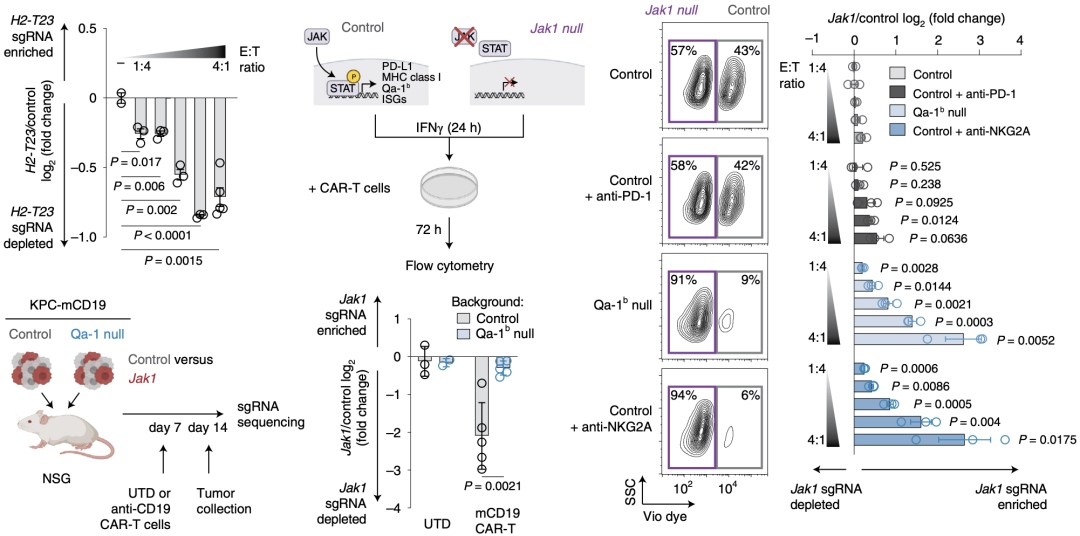
IFN信号通过上调 Qa-1b来抑制 CAR-T 细胞毒性
最后,Manguso团队探讨了IFN信号对于免疫治疗响应预测的能力。
他们分析了一组接受抗PD-1单抗治疗的黑色素瘤患者队列(6p21.3两倍性),发现ISG的表达与更好的响应相关,这一结果与前述的ccRCC患者队列中的结果相反。
这提示利用IFN特征预测ICB响应需要考虑CD8+T细胞和NK细胞的响应优势,在NK细胞响应占优时,IFN特征与接受抗PD-1治疗差的响应相关;而在CD8+T细胞响应占优时,IFN特征与接受抗PD-1治疗好的响应相关。
高IFN特征在不同肿瘤微环境中与不同的ICB响应相关
Manguso团队还评估了1个接受ICB治疗的202名黑色素瘤患者的队列,分别在基线、治疗后6周和治疗后6个月时间点取样,检测血清中ISG蛋白的水平。
结果显示,在治疗后6周时,响应和无响应患者血清ISG蛋白含量都会增加。而治疗后6个月时,无响应患者血清ISG蛋白含量显著增加。并且,在这2个时间点,血清ISG的变化都与患者更差的无进展生存相关。因此,接受ICB治疗过程中,对于ICB的抵抗可能与IFN和ISG的水平升高有关。
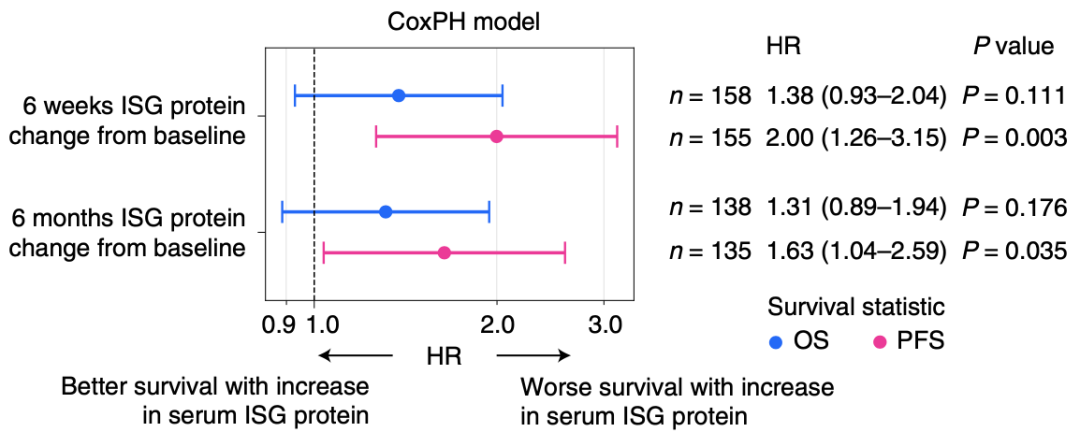
ICB治疗后血清ISG蛋白水平升高与治疗抵抗有关
总的来说,这项研究发现了IFN感应性的丧失在多个动物模型中使得肿瘤对ICB更加敏感。这可以通过两种机制介导,第一是经典MHC-I类分子的上调能够抑制NK细胞,第二是非经典MHC-I类分子Qa-1b的上调可以通过NKG2A/CD94抑制CD8+T细胞。
既往研究往往关注IFN感应性的丧失会引起对ICB的抵抗,这项研究提示了IFN感应性功能的复杂性,IFN介导的MHC-I类分子上调可以发挥促进对CD8+T细胞抗原呈递和抑制NK细胞的双重作用。在高表达NK配体(MICA和MICB)的肿瘤中,由于IFN驱动的MHC-I类分子上调可以抑制NK细胞,高ISG特征与差的响应相关。而在T细胞高度克隆扩增的肿瘤中,由于IFN能够促进对T细胞的抗原呈递,高ISG特征与好的响应相关。因此,ISG特征在肿瘤中的作用可能取决于肿瘤中的免疫效应主要由NK细胞还是CD8+T细胞介导。
除此之外,这项研究中发现非经典MHC-I类分子Qa-1b(HLA-E)可以发挥免疫抑制作用,非经典MHC-I类分子如HLA-F、HLA-G可能发挥的免疫抑制作用也值得进一步探索。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
