
CD:肺癌为何会对一代又一代的EGFR-TKI耐药?这个研究破案了!
来源:奇点糕 2022-11-01 17:29
全球每年约有220万人罹患肺癌,180万人死于肺癌[1],肺癌严重威胁人类生命健康。EGFR突变是最常见的肺癌突变类型,尤其在包括中国在内的亚洲地区。
全球每年约有220万人罹患肺癌,180万人死于肺癌[1],肺癌严重威胁人类生命健康。EGFR突变是最常见的肺癌突变类型,尤其在包括中国在内的亚洲地区。
自2002年问世以来,针对EGFR突变的酪氨酸激酶抑制剂(EGFR-TKI),大大延长了肺癌患者的生存时间,已成为EGFR突变肺癌一线治疗的最佳选择。
可是,道高一尺,魔高一丈。EGFR-TKI应用一段时间后,患者不可避免地出现耐药,成为制约肺癌靶向治疗的一个难点[2]。为克服耐药,EGFR-TKI不断更新换代,“打补丁”式地进行完善,从以吉非替尼为代表的一代,到以奥希替尼为代表的三代,再到临床试验中的四代,一代更比一代强,然而,耐药依旧层出不穷。
治疗过程中产生新突变,是肺癌获得性耐药的一个重要原因[3]。如对一代和二代TKI耐药的T790M突变,对三代TKI耐药的C797S突变。但是,其详细机制尚有待阐明。
近日,由以色列魏茨曼科学研究所Yosef Yarden领衔的研究团队,揭示了肺癌经EGFR-TKI治疗产生耐药突变的机制,相关论文发表于肿瘤学顶级期刊Cancer Discovery上[4]。
他们发现,肺癌细胞经EGFR-TKI刺激后,AXL被激活。AXL一方面激活RAD18,启动DNA易错旁路修复机制,加速耐药突变的出现;另一方面激活MYC,调控嘌呤合成,使核苷酸库失衡,增强诱变。如果在EGFR-TKI的治疗中联用抗AXL抗体,能抑制耐药性的产生,防止肺癌复发。

官网论文首页截图
研究伊始,研究人员使用EGFR-TKI处理肺癌细胞,获得不同程度的耐药亚克隆细胞,对这些细胞进行转录组测序,发现GAS6是持久耐药细胞中最明显的高表达基因。与此同时,来自临床的样本也发现,EGFR-TKI治疗失败的肺癌患者也高表达GAS6。
既往研究发现,GAS6及其受体AXL在癌症中参与了肿瘤细胞的增殖和转移[5]。研究人员对AXL在肺癌中的表达进行检测,发现TKI治疗后复发的肺癌小鼠AXL表达升高,AXL磷酸化水平也上调。这提示GAS6-AXL轴可能还参与肺癌的TKI耐药。
为了检验假说,研究人员分别过表达和敲除肺癌细胞的AXL,然后把细胞暴露于EGFR-TKI。相比于野生型的肺癌细胞,AXL过表达细胞存活率增加,而AXL敲除细胞存活率降低。
接着,研究人员又在体内对AXL的促耐药作用进行了验证,把AXL过表达肺癌细胞和AXL敲除细胞分别接种于裸鼠,然后用TKI治疗荷瘤小鼠。AXL过表达的肺癌小鼠更早地出现耐药,AXL敲除的肺癌小鼠则完全没有复发现象。
这些结果表明,GAS6-AXL轴被TKI治疗激活后,促进了肺癌细胞耐药。
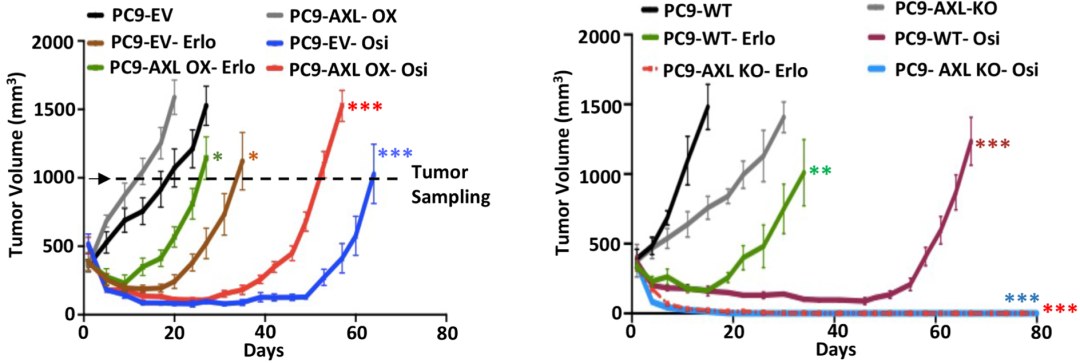
AXL过表达的肺癌小鼠生长加快,AXL敲除的肺癌小鼠肿瘤生长减慢
我们都知道,DNA损伤是对细胞的致命打击,为应对DNA损伤,细胞会启动DNA修复机制。TLS聚合酶参与DNA损伤修复,直接在损伤的DNA上掺入核苷酸,延续DNA复制。而TKI的作用机制之一即是破坏细胞对DNA损伤的修复能力[6]。
于是,研究人员检测了TKI治疗后肺癌细胞的DNA损伤和修复情况。
经TKI处理后,肺癌细胞DNA双链断裂,产生ROS,凋亡增加,存活下降,RAD18和TLS聚合酶等DNA修复相关酶表达上调。而在AXL过表达后,肺癌细胞能抵抗TKI的打击,DNA断裂减少,ROS水平降低,凋亡被抑制,存活率升高,RAD18和TLS聚合酶的表达上调至更高水平。
RAD18是一种E3泛素连接酶,通过诱导PCNA单泛素化和招募TLS聚合酶来激活DNA的易错旁路修复[7]。易错旁路途径虽然能提高细胞对DNA损伤的耐受性,但却牺牲了复制的保真度,它在损伤的DNA模板对侧掺入错误的核苷酸,引发基因组突变。
研究人员对AXL和RAD18的关系进行了探究,发现AXL能与RAD18结合,增强RAD18的类泛素化,减少RAD18的泛素化,因而增强RAD18对PCNA的单泛素化修饰,进而启动易错旁路途径。
接着,研究人员检测了TKI治疗后肺癌的基因组变化情况,发现TKI诱导了DNA的碱基改变,耐药突变T790M开始出现。AXL的表达水平和肺癌的突变负担呈正相关,过表达AXL能增加T790M突变频率,使用抗AXL抗体处理肺癌细胞,可显著降低T790M突变的丰度。
这些结果表明,AXL促进了TKI对肺癌诱导的RAD18和TLS聚合酶表达上调,激活了易错旁路途径,加速了耐药突变的产生。
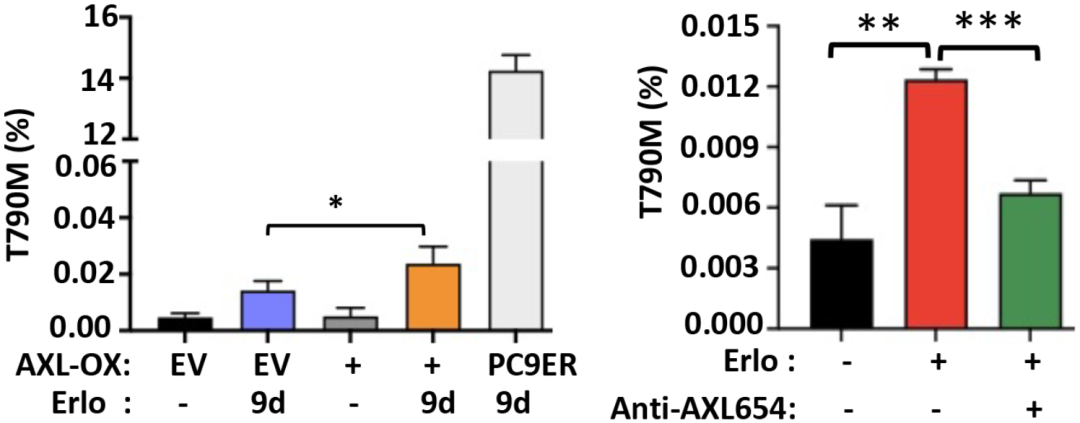
AXL能提高T790M耐药突变频率,抗AXL抗体可显著降低T790M突变
对AXL调控的通路进行分析,研究人员发现AXL能影响肺癌细胞的核苷酸代谢,主要是嘌呤、组氨酸和谷氨酰胺途径。由于DNA的复制取决于脱氧核苷酸池[8],研究人员推测,AXL可能还通过核苷酸代谢,进一步增强诱变作用。
研究人员分别过表达和敲除AXL,发现肺癌细胞嘌呤代谢相关基因的表达,以及嘌呤合成途径中的代谢物也相应地发生变化。同位素标记实验显示,AXL加速了嘌呤的合成。
MYC是AXL的转录靶点,在肺癌中,MYC的表达和AXL呈正相关,而MYC能调控嘌呤合成酶。因此,研究人员得出结论,AXL通过激活MYC促进嘌呤合成,进而增强肺癌的获得性突变。
最后,研究人员开发出了一种抗AXL抗体,分别在体内外检测抑制AXL对TKI疗效的增强作用。
在EGFR-TKI的治疗中联用抗AXL抗体,肺癌细胞的AXL、RAD18和TLS聚合酶表达下调,凋亡增加,耐药细胞减少。动物实验表明,相比于单药治疗后出现复发,肺癌小鼠在联合用药后完全无复发。抗AXL抗体能抑制肺癌的TKI耐药,消除复发。
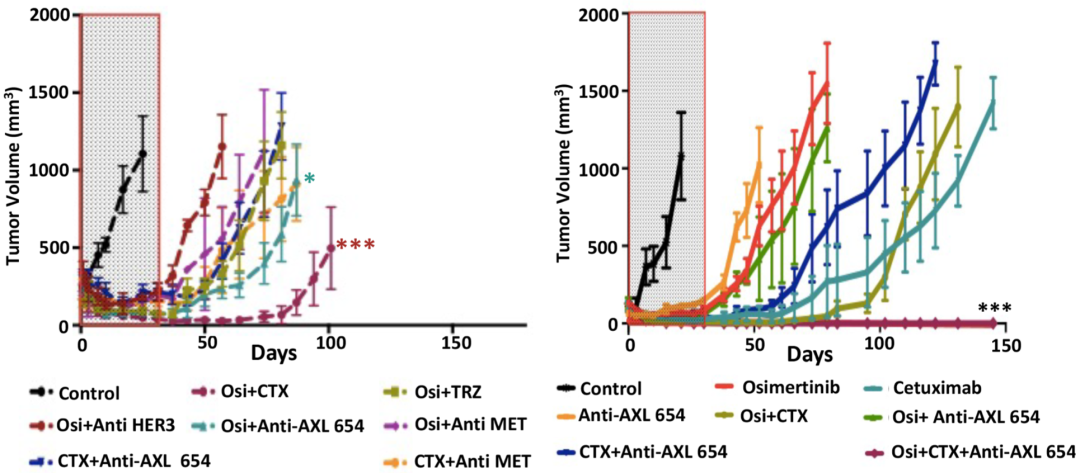
EGFR-TKI治疗联用抗AXL抗体能抑制肺癌生长,防止复发
总的来说,研究人员发现暴露于EGFR-TKI后,肺癌细胞激活了GAS6-AXL途径,AXL同时通过非转录和转录这两种方式促进耐药突变的产生。非转录方式通过增强类泛素化和减少泛素化激活RAD18,RAD18招募TLS聚合酶启动易错旁路途径。转录方式通过激活MYC促进嘌呤合成,进而使肺癌细胞的核苷酸库失衡。
这项研究不仅揭示了肺癌在EGFR-TKI治疗后出现耐药的机制,也用实验证明靶向AXL是消除EGFR突变型肺癌耐药的一个潜在策略。
参考文献
[1] Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209-249.
[2] Nagano T, Tachihara M, Nishimura Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells. 2018;7(11):212. Published 2018 Nov 15. doi:10.3390/cells7110212
[3] Garraway LA, Jänne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2(3):214-226. doi:10.1158/2159-8290.CD-12-0012
[4] Noronha A, Belugali Nataraj N, Sang Lee J, et al. AXL and error-prone DNA replication confer drug resistance and offer strategies to treat EGFR-mutant lung cancer [published online ahead of print, 2022 Jul 27]. Cancer Discov. 2022;CD-22-0111. doi:10.1158/2159-8290.CD-22-0111
[5] Wu G, Ma Z, Hu W, et al. Molecular insights of Gas6/TAM in cancer development and therapy. Cell Death Dis. 2017;8(3):e2700. Published 2017 Mar 23. doi:10.1038/cddis.2017.113
[6] Liang XM, Qin Q, Liu BN, et al. Targeting DNA-PK overcomes acquired resistance to third-generation EGFR-TKI osimertinib in non-small-cell lung cancer. Acta Pharmacol Sin. 2021;42(4):648-654. doi:10.1038/s41401-020-00577-1
[7] Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425(6954):188-191. doi:10.1038/nature01965
[8] Schmidt TT, Reyes G, Gries K, et al. Alterations in cellular metabolism triggered by URA7 or GLN3 inactivation cause imbalanced dNTP pools and increased mutagenesis. Proc Natl Acad Sci U S A. 2017;114(22):E4442-E4451. doi:10.1073/pnas.1618714114
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
