
研究揭示CDKL5缺乏症癫痫发生发展的分子细胞机制与干预靶点
来源:脑智卓越中心 2023-10-08 09:55
熊志奇团队致力于推动CDKL5缺乏症的基础与临床研究,并与患儿家长发起成立了中国CDKL5互助联盟和希舞关爱之家。
Cell Reports在线发表了题为CDKL5 deficiency in adult glutamatergic neurons alters synaptic activity and causes spontaneous seizures via TrkB signaling的研究论文。该研究由中国科学院脑科学与智能技术卓越创新中心、上海脑科学与类脑研究中心研究员熊志奇研究组和上海交通大学医学院附属精神卫生中心朱永川研究组合作完成。
CDKL5缺乏症(CDKL5 deficiency disorder,CDD)是由位于X染色体的CDKL5基因功能缺失性突变导致的神经发育疾病。CDD主要症状包括早发性癫痫、刻板动作以及认知与运动障碍等。2003年,中国学者陶炯与合作导师德国马普分子遗传研究所教授Vera Kalscheuer在智力障碍伴婴儿痉挛患者中发现了CDKL5基因的突变。2004年,陶炯与熊志奇合作开启了CDKL5的功能研究,克隆了啮齿类动物的同源基因,揭示了CDKL5在大脑中的时空表达特征,并在2010年报道了CDKL5在神经元形态发育中的作用与机制。
早发性难治性癫痫发作是CDD患儿的典型症状。这一疾病首次发作通常出现在出生后的3个月内,给患者和家庭带来身心负担。早期的CDD动物模型,如Cdkl5基因全敲除以及致病突变敲入小鼠,均未表现出癫痫发作。2021年,熊志奇研究组发现,在出生早期条件性敲除前脑兴奋性神经元中的Cdkl5基因可致小鼠自发性癫痫发作,这改变了当时领域内普遍的观点即CDD模型小鼠无法体现类似临床的癫痫症状。尽管科研人员在CDD小鼠模型中重现了癫痫发作的表型,但这些小鼠存在发作时间延迟、进程慢等问题。本研究发现,在成年小鼠中特异性敲除前脑兴奋性神经元中的Cdkl5基因能够使所有小鼠在两周后出现自发性癫痫发作,而在发育期诱导敲除Cdkl5基因则发作癫痫的小鼠比例下降。这表明小鼠脑发育过程中的代偿效应可能是影响出现癫痫表型的重要原因,绕过发育过程在成年大脑失活CDKL5是建立CDD癫痫模型的关键。
研究利用新构建的CDD癫痫小鼠模型发现,脑源性神经营养因子(brain-derived neurotrophic factor, BDNF)的表达在癫痫发作前增加,其受体TrkB及下游信号分子亦存在过度激活,这导致海马齿状回突触前囊泡释放概率增大、兴奋性突触传递增强。利用遗传学手段敲除一个拷贝的TrkB编码基因来降低TrkB的表达,能够阻止CDD小鼠的癫痫发生与发展。更具有临床意义的是,在已发作癫痫的小鼠中,通过药理学手段抑制TrkB受体的活性显著减少了癫痫发作。这表明BDNF-TrkB信号通路在CDD癫痫小鼠模型里的异常激活是导致兴奋性突触传递增强和癫痫发作的重要机制。
熊志奇团队致力于推动CDKL5缺乏症的基础与临床研究,并与患儿家长发起成立了中国CDKL5互助联盟和希舞关爱之家。该团队揭示了CDKL5在神经元迁移和树突/轴突生长中的重要作用、CDKL5在兴奋性突触中的作用和机制以及CDKL5自身磷酸化在神经元发育和可塑性中的功能,并在国际上首次构建了具有显著自发癫痫表型的CDD小鼠模型,发现了潜在的药物干预靶点。
研究工作获得中国科学院、科学技术部、国家自然科学基金委员会和上海市的资助,并得到脑智卓越中心光学成像平台、分子细胞技术平台和实验动物平台的技术支持。
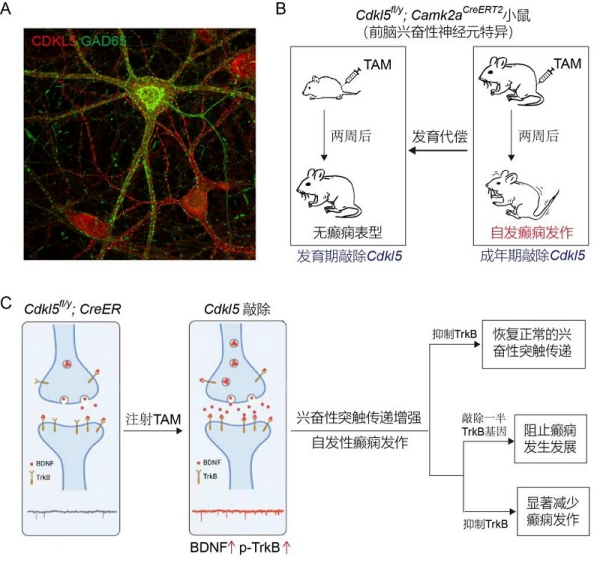
CDKL5功能缺失通过BDNF-TrkB信号通路引起自发性癫痫发作。A、CDKL5(红色)在兴奋性神经元和GAD65阳性抑制性神经元(绿色)中表达。B、成年小鼠前脑兴奋性神经元中敲除Cdkl5基因引起自发性癫痫发作。C、BDNF-TrkB参与CKDL5功能缺失引起的兴奋性突触增强和自发性癫痫发作。敲除一半的TrkB编码基因能够阻止癫痫的发生和发展;而在癫痫发作开始后抑制TrkB活性能够显著减少癫痫发作。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
