
全文编译!复旦大学在Cell期刊上发文,从基因组学角度揭示SARS-CoV-2的起源和出现
来源:本站原创 2020-03-31 11:09
2020年3月31日讯/生物谷BIOON/---新型人类冠状病毒SARS-CoV-2(之前称为2019-nCoV)的持续大流行引起了全球的极大关注。我们和中国的其他人参与了对这种病毒的初始基因组测序。在本文中,我们描述了针对SARS-CoV-2的出现,这些基因组数据揭示了什么,并讨论了我们对其起源理解上的差距。一种新的人类冠状病毒2019年12月下旬在中国湖
2020年3月31日讯/生物谷BIOON/---新型人类冠状病毒SARS-CoV-2(之前称为2019-nCoV)的持续大流行引起了全球的极大关注。我们和中国的其他人参与了对这种病毒的初始基因组测序。在本文中,我们描述了针对SARS-CoV-2的出现,这些基因组数据揭示了什么,并讨论了我们对其起源理解上所存在的差距。
一种新的人类冠状病毒
2019年12月下旬在中国湖北省武汉市出现了首批关于新型肺炎(COVID-19)病例的报告,不过回顾性分析已确认一名患者早在2019年12月1日就出现了症状。鉴于SARS-CoV-2病例的数量正在快速增长并在全球范围内蔓延,我们将避免引用已确诊的感染病例数。然而,实际病例数可能会大大超过报告的病例数,这是因为非常轻微或无症状的感染常被排除在计数之外。任何病例数少报的情况显然意味着,在疫情最严重的地区,与COVID-19相关的病死率(case fatality rate, CFR)将低于目前引用的数字。病死率还将在地理上、年龄组之间和时间上有所不同。尽管没有大规模的血清学调查可能无法解决这些不确定性,但是从当前数据来看,很明显,COVID-19的病死率显著高于季节性流感,但低于最近在人类中出现的两种密切相关的冠状病毒---导致2002/2003年严重急性呼吸道综合征(SARS)疫情爆发的SARS-CoV;MERS-CoV,自2015年以来,它一直是中东呼吸综合征(MERS)疫情主要集中在阿拉伯半岛爆发的主要原因---的病死率。但是,也很明显,SARS-CoV-2的传染性比SARS-CoV和MERS-CoV都高,而且患者可在无症状时或有症状前传播这种病毒,但传播的频率仍不确定。
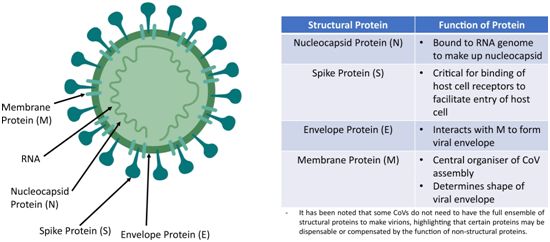
考虑到SARS-CoV-2无疑有一个人畜共患病的起源,它与一家海鲜市场之间存在的关联性应当是不足为奇的。然而,鉴于并非所有早期病例都与这家海鲜市场相关,因此SARS-CoV-2出现的故事可能比最初想象的更为复杂。如今从这家海鲜市场上获得了“环境样本”(可能是物体表面)的基因组序列,而且针对它们的系统进化树分析显示它们与从最早的武汉患者中采样的病毒密切相关。尽管这再次表明这家海鲜市场在病毒出现中发挥了重要作用,但尚不清楚这些样本是否来自无意中获得了传染性物质的人还是源自这个地方的动物或动物残留物。不幸的是,这家海鲜市场上明显缺乏直接的动物采样,这可能意味着,很难甚至不可能准确地识别出这个地方的任何动物宿主。
在临床病例开始出现之后,我们的研究团队以及其他一些团队尝试确定这种致病病原体的基因组序列。我们着重关注是一名在症状发作六天后于2019年12月26日在武汉市中心医院住院的患者。这名患者正出现发烧、胸闷、咳嗽、疼痛和无力,以及指示肺炎的肺部异常,其中这些肺部异常似乎在COVID-19中很常见。幸运的是,下一代宏转录组测序使得我们能够在2020年1月5日从这名患者体内获得完整的病毒基因组。初步分析显示这种病毒与SARS样冠状病毒(冠状病毒科)密切相关。
这一结果立即报告给有关部门,并在同一天将这个基因组序列(Wuhan-Hu-1毒株)的注释版本提交给NCBI/GenBank。尽管GenBank序列(登录号MN908947)是SARS-CoV-2的第一个可用序列,但随后对它进行了校正以确保其准确性。
在英国爱丁堡大学Andrew Rambaut博士的帮助下,我们于2020年1月11日(格林威治标准时间, GMT)早些时候在开放存取的virological.org网站(http://virological.org/)上发布了这种病毒的基因组序列。不久之后,中国疾控中心在公众可访问的GISAID数据库(https://www.gisaid.org/)上类似地发布了SARS-CoV-2基因组序列(以及相关的流行病学数据)。在撰写本文时,已有近200个SARS-CoV-2基因组可公开获得,这代表了来自中国及其他地区的这种病毒的基因组多样性,并提供了可自由获取的全球资源。重要的是,SARS-CoV-2基因组序列数据的发布促进了诊断测试和感染性克隆(infectious clone)的快速开发。研发有效疫苗和抗病毒药物的竞赛正在进行中,而且针对抗病毒药物的临床试验正在逐步展开。
比较SARS-CoV-2和其他的冠状病毒
这些最早的基因组序列数据清楚地表明SARS-CoV-2是乙型冠状病毒属(Betacoronavirus,有时也译为β冠状病毒属)的成员,属于一个包括SARS-CoV在内的Sarbecovirus亚属(MERS-CoV属于一个单独的亚属:Merbecovirus)。确实,初步比较显示SARS-CoV-2在核苷酸水平上与SARS-CoV的相似度约为79%。当然,在不同基因上的相似性模式差异很大:SARS-CoV和SARS-CoV-2在刺突蛋白(S)基因上仅显示约72%的核苷酸序列相似性,其中S蛋白是一种与宿主细胞受体相互作用的关键表面糖蛋白。
考虑到这些密切的进化关系,SARS-CoV-2的基因组结构类似于其他β冠状病毒就不足为奇了,它的基因顺序为5'-复制酶ORF1ab-S-envelope(E)-membrane(M)-N-3'。SARS-CoV-2的较长复制酶ORF1ab基因长度超过21 kb,包含16个预测的非结构蛋白以及许多在功能上可能与SARS-CoV相类似的下游开放阅读框(ORF)。2013年从中国云南省采集的中华菊头蝠中获得的一种相关病毒极大地促进了比较基因组分析。这种病毒称为RaTG13,在核苷酸序列水平上与SARS-CoV-2的相似度约为96%。尽管有这种序列相似性,但是SARS-CoV-2和RaTG13在许多关键基因组特征上还是不同的,可以说,其中最重要的是SARS-CoV-2在S蛋白的S1亚基和S2亚基交界处含有一个多碱性切割位点(polybasic cleavage site)插入。尽管位于S1/S2切割位点附近的多碱性位点存在于包括HCoV-HKU1在内的其他人类冠状病毒和高致病性的禽流感病毒毒株中,但是这种可能增加了SARS-CoV-2传染性的插入并不存在于相关的蝙蝠β冠状病毒中。此外,SARS-CoV-2和RaTG13的受体结合结构域(RBD)仅有大约85%的相似度,并且在RBD的六个关键氨基酸残基中仅有一个是相同的。重要的是,序列和结构比较都提示着SARS-CoV-2 RBD非常适合与也被SARS-CoV使用的人ACE2受体结合。重要的是,最近在2019年中期从云南省另一种菊头蝠中取样的病毒(RmYN02)中观察到了在S1/S2切割位点上氨基酸PAA的独立插入,这表明这些插入事件反映了冠状病毒正在进行的进化的一个自然部分。虽然RmYN02在S蛋白上与SARS CoV-2相对不同(?72%序列相似度),但是在较长的复制酶基因上,它与这种人类冠状病毒具有最为密切的亲缘关系(?97%的核苷酸序列相似度)。
尽管SARS-CoV和MERS-CoV与SARS-CoV-2密切相关并且都有蝙蝠宿主,但这两种病毒之间的生物学差异却很明显。如上所述,SARS-CoV-2的传染性明显更高,从而导致与SARS-CoV和MERS-CoV完全不同的流行病学动态。在后两种病毒中,病例数的增长相对较慢,而MERS-CoV从未完全适应人类传播:大多数病例是由于阿拉伯半岛的骆驼溢出造成的,仅零星地发生人与人之间的传播。相比之下,SARS-CoV-2在当地的显著传播最让人吃惊。确定支撑这种传播能力的病毒学特征显然是当务之急。
SARS-CoV-2的人畜共患病起源
COVID-19的出现和迅速传播标志着一场完美的流行病风暴。这是一种毒性相对较高的呼吸道病原体,它来自一个具有跨越物种界限的不寻常本领的病毒家族。它出现在一个主要的人口中心和旅游中心,就在一年中最大的旅游旺季---中国春节---前夕。确实,流行病学模型表明SARS-CoV-2在武汉市被严格隔离之前已经在中国广泛传播也就不足为奇了。
早期基因组比较显示这种与SARS-CoV关系最密切的病毒也来自蝙蝠,这也不足为奇。近年来的采样已经确定了一系列令人印象深刻的蝙蝠冠状病毒,包括RaTG13和RmYN02。因此,对于多种冠状病毒而言,蝙蝠无疑是重要的宿主。尽管如此,蝙蝠在SARS-CoV-2的人畜共患病起源中所起的确切作用尚未确定。特别是,这些与SARS-CoV-2关系最密切的蝙蝠冠状病毒是从距武汉市1500多公里的云南省的动物中取样的。湖北省的蝙蝠冠状病毒相对较少,而且那些已被测序的蝙蝠冠状病毒在系统进化树上与SARS-CoV-2距离相对较远。基于此的一个简单的推断是,我们对蝙蝠病毒的采样强烈偏向某些地理位置。这将需要在以后的研究中加以纠正。此外,尽管96%~97%的序列相似度听起来像是现有的蝙蝠病毒与SARS-CoV-2密切相关,但是实际上这可能代表20多年的序列进化(然而,如果这种病毒在人类中有很强的适应性进化,那么潜在的分子时钟可能以不确定的速度滴答作响)。因此,几乎可以肯定的是,更多的采样将会鉴定出与SARS-CoV-2亲缘关系更近的其他蝙蝠病毒。一个关键问题是这些病毒或来自任何其他动物的病毒是否包含这些关键的RBD突变以及与SARS-CoV-2中相同的弗林蛋白酶样切割位点插入。
尽管蝙蝠很可能是这种病毒的天然宿主,但它们与人类之间的生态隔离使得其他哺乳动物物种很可能充当“中间”或“扩增”宿主,在这些中间宿主中,SARS-CoV-2能够获得在人类中有效传播所需的部分或全部突变。就SARS-CoV和MERS-CoV而言,果子狸和骆驼分别起着中间宿主的作用,尽管MERS-CoV可能在骆驼体内存在了几十年,然后才在人类身上出现,但在多次跨物种事件中,这些动物可能被更好地认为是真正的宿主。为了确定这些中间宿主物种可能是什么,必须对来自湿货市场或居住在人群附近的动物进行更广泛的采样。最近在非法进口到中国南部(广东和广西)的马来亚穿山甲中发现了与SARS-CoV-2密切相关的病毒,这突出说明了这一点。广东穿山甲冠状病毒在RBD上与SARS-CoV-2特别密切相关,包含认为使得与ACE2受体结合的六个关键突变中的全部六个(尽管它们在基因组的剩余部分上与SARS-CoV-2存在更大的差异)。尽管穿山甲因经常被人们非法贩运和处于濒危状态而引起人们极大的兴趣,但是穿山甲携带与SARS-CoV-2相关的病毒强烈表明,各种哺乳动物中都存在大量相关的β冠状病毒但尚未取样。 虽然我们过去在冠状病毒中的经验表明,动物宿主---天然宿主和中间宿主---的进化解释人类SARS-CoV-2出现的必要条件,但是不能排除这种病毒在2019年12月首次被发现之前在一段‘隐性’传播时间内在人类中获得了一些关键突变。特别是,这种病毒可能比预期的更早在人群中出现(也许甚至最早并不在武汉出现),但是由于无症状感染病例,具有轻度呼吸道症状的病例,甚至零星的肺炎病例,它在用于监视和病原体识别的标准系统中并未被观察到。在这段隐性传播期间,这种病毒可能逐渐获得了关键突变(可能包括RBD和弗林蛋白酶切割位点插入),从而使得它能够完全适应人类。直到发生了一系列肺炎病例,我们才能够通过常规监视系统检测到COVID-19。显然,对呼吸道感染的回顾性血清学或宏基因组学研究将有助于确定这种情况是否正确,尽管这样的早期病毒可能永远不会被检测到。
另一个受到广泛关注的问题是SARS-CoV-2是否是一种重组病毒,以及这种重组是否可能促进了它的出现。一个复杂的因素是sarbevirus病毒和更广泛的冠状病毒经历了广泛的重组,因此区分有助于病毒出现的重组与“背景”重组事件并非易事。重组可见于sarbevirus病毒基因组的多个位置(包括在S蛋白中)以及与SARS-CoV-2密切相关的蝙蝠病毒中。比如,有证据表明SARS-CoV-2、RaTG13和广东穿山甲冠状病毒之间会发生重组,而RmYN02的基因组类似地受到重组的影响。但是,很难确定重组事件的确切模式和基因组谱系,尤其是因为许多重组区域可能较小,并且随着我们对更多与SARS-CoV-2相关的病毒进行采样,这些重组区域可能会发生变化。为了解决这些问题,再次有必要对动物种群中的病毒多样性进行更广泛的采样。
正在进行的SARS-CoV-2基因组进化
随着COVID-19流行病的发展,更多的病毒基因组已被测序。正如基于它们最近的共同祖先所预期的那样,来自武汉的最早样本包含相对较少的遗传多样性。虽然这可以阻止详细的系统进化树和谱系地理学推断,但是它确实表明武汉的公共卫生部门在检测第一批肺炎聚集性病例方面做得非常出色。但是,这种看似最近的共同祖先并不排除存在一个在人类中隐性传播的疫情爆发前时期。
尽管积累遗传多样性意味着如今有可能检测到SARS-CoV-2序列的不同系统进化树簇,但仅通过基因组比较很难确定这种病毒在全球人群中传播时是否固定了表型上重要的突变,而且任何此类声明都需要仔细的实验验证。考虑到RNA病毒特征性的高突变率,很明显在这种病毒基因组中将出现更多的突变,这些突变将有助于我们追踪SARS-CoV-2的传播。
但是,随着流行病的蔓延,我们的序列样本量相对于病例总数来说可能非常小,以至于很难(如果不是不可能的话)检测每个传播链。因此,在尝试推断确切的传播事件时必须格外小心。顺便说一句,尽管冠状病毒的突变率可能比其他RNA病毒低,这是由于3'→5'核糖核酸外切酶内在地具有一定的校对活性,但它们的长期核苷酸替换速率(即分子进化)分布与其他RNA病毒相同。这提示着较低的突变率在一定程度上被宿主内较高的病毒复制速率所补偿。尽管没有证据表明这种突变能力(对RNA病毒常见)会导致表型---比如传播性和毒力---发生任何根本性变化,这是因为这些变化很少在单个疾病疫情规模上发生变化,但是监视病毒传播时表型的任何变化显然很重要。很有可能,COVID-19的病例数和/或病死率的任何下降都可能是由于人群免疫力的提高和流行病学背景而不是这种病毒中的突变变化引起的。
结论
SARS-CoV-2似乎将不可避免地成为人类中的第五种地方性冠状病毒(其他四种为HCoV-OC43、HCoV-229E、HCoV-NL63和HCoV-HKU1),并且目前正在完全易感人群中传播。冠状病毒显然具有跨越物种界限并适应新宿主的能力,这使得预测未来会有更多的病毒出现变得很简单,不过尚不清楚为何相比于一些其他的RNA病毒,冠状病毒具有这种能力。至关重要的是,对动物冠状病毒的监视应包括蝙蝠以外的动物,这是因为中间宿主的作用可能非常重要,从而为病毒在人类中的出现提供了更直接的途径。
鉴于野生动物中病毒的多样性以及它们的持续进化,可以说,降低未来疫情爆发风险的最简单最具成本效益的方法是尽可能限制我们对动物病原体的接触。尽管我们与动物世界的亲密关系意味着我们无法建立坚不可摧的屏障,但是针对非法野生动物贸易采取更强有力的行动,以及将所有野生哺乳动物(也许还有禽类)从湿货市场中清除将提供一个重要的缓冲。(生物谷 Bioon.com)
参考资料:
Yong-Zhen Zhang et al. A Genomic Perspective on The Origin and Emergence of SARS-CoV-2. Cell, 2020, doi: 10.1016/j.cell.2020.03.035.
一种新的人类冠状病毒
2019年12月下旬在中国湖北省武汉市出现了首批关于新型肺炎(COVID-19)病例的报告,不过回顾性分析已确认一名患者早在2019年12月1日就出现了症状。鉴于SARS-CoV-2病例的数量正在快速增长并在全球范围内蔓延,我们将避免引用已确诊的感染病例数。然而,实际病例数可能会大大超过报告的病例数,这是因为非常轻微或无症状的感染常被排除在计数之外。任何病例数少报的情况显然意味着,在疫情最严重的地区,与COVID-19相关的病死率(case fatality rate, CFR)将低于目前引用的数字。病死率还将在地理上、年龄组之间和时间上有所不同。尽管没有大规模的血清学调查可能无法解决这些不确定性,但是从当前数据来看,很明显,COVID-19的病死率显著高于季节性流感,但低于最近在人类中出现的两种密切相关的冠状病毒---导致2002/2003年严重急性呼吸道综合征(SARS)疫情爆发的SARS-CoV;MERS-CoV,自2015年以来,它一直是中东呼吸综合征(MERS)疫情主要集中在阿拉伯半岛爆发的主要原因---的病死率。但是,也很明显,SARS-CoV-2的传染性比SARS-CoV和MERS-CoV都高,而且患者可在无症状时或有症状前传播这种病毒,但传播的频率仍不确定。
图片来自Eye, 2020, doi:10.1038/s41433-020-0790-7。
考虑到SARS-CoV-2无疑有一个人畜共患病的起源,它与一家海鲜市场之间存在的关联性应当是不足为奇的。然而,鉴于并非所有早期病例都与这家海鲜市场相关,因此SARS-CoV-2出现的故事可能比最初想象的更为复杂。如今从这家海鲜市场上获得了“环境样本”(可能是物体表面)的基因组序列,而且针对它们的系统进化树分析显示它们与从最早的武汉患者中采样的病毒密切相关。尽管这再次表明这家海鲜市场在病毒出现中发挥了重要作用,但尚不清楚这些样本是否来自无意中获得了传染性物质的人还是源自这个地方的动物或动物残留物。不幸的是,这家海鲜市场上明显缺乏直接的动物采样,这可能意味着,很难甚至不可能准确地识别出这个地方的任何动物宿主。
在临床病例开始出现之后,我们的研究团队以及其他一些团队尝试确定这种致病病原体的基因组序列。我们着重关注是一名在症状发作六天后于2019年12月26日在武汉市中心医院住院的患者。这名患者正出现发烧、胸闷、咳嗽、疼痛和无力,以及指示肺炎的肺部异常,其中这些肺部异常似乎在COVID-19中很常见。幸运的是,下一代宏转录组测序使得我们能够在2020年1月5日从这名患者体内获得完整的病毒基因组。初步分析显示这种病毒与SARS样冠状病毒(冠状病毒科)密切相关。
这一结果立即报告给有关部门,并在同一天将这个基因组序列(Wuhan-Hu-1毒株)的注释版本提交给NCBI/GenBank。尽管GenBank序列(登录号MN908947)是SARS-CoV-2的第一个可用序列,但随后对它进行了校正以确保其准确性。
在英国爱丁堡大学Andrew Rambaut博士的帮助下,我们于2020年1月11日(格林威治标准时间, GMT)早些时候在开放存取的virological.org网站(http://virological.org/)上发布了这种病毒的基因组序列。不久之后,中国疾控中心在公众可访问的GISAID数据库(https://www.gisaid.org/)上类似地发布了SARS-CoV-2基因组序列(以及相关的流行病学数据)。在撰写本文时,已有近200个SARS-CoV-2基因组可公开获得,这代表了来自中国及其他地区的这种病毒的基因组多样性,并提供了可自由获取的全球资源。重要的是,SARS-CoV-2基因组序列数据的发布促进了诊断测试和感染性克隆(infectious clone)的快速开发。研发有效疫苗和抗病毒药物的竞赛正在进行中,而且针对抗病毒药物的临床试验正在逐步展开。
比较SARS-CoV-2和其他的冠状病毒
这些最早的基因组序列数据清楚地表明SARS-CoV-2是乙型冠状病毒属(Betacoronavirus,有时也译为β冠状病毒属)的成员,属于一个包括SARS-CoV在内的Sarbecovirus亚属(MERS-CoV属于一个单独的亚属:Merbecovirus)。确实,初步比较显示SARS-CoV-2在核苷酸水平上与SARS-CoV的相似度约为79%。当然,在不同基因上的相似性模式差异很大:SARS-CoV和SARS-CoV-2在刺突蛋白(S)基因上仅显示约72%的核苷酸序列相似性,其中S蛋白是一种与宿主细胞受体相互作用的关键表面糖蛋白。
考虑到这些密切的进化关系,SARS-CoV-2的基因组结构类似于其他β冠状病毒就不足为奇了,它的基因顺序为5'-复制酶ORF1ab-S-envelope(E)-membrane(M)-N-3'。SARS-CoV-2的较长复制酶ORF1ab基因长度超过21 kb,包含16个预测的非结构蛋白以及许多在功能上可能与SARS-CoV相类似的下游开放阅读框(ORF)。2013年从中国云南省采集的中华菊头蝠中获得的一种相关病毒极大地促进了比较基因组分析。这种病毒称为RaTG13,在核苷酸序列水平上与SARS-CoV-2的相似度约为96%。尽管有这种序列相似性,但是SARS-CoV-2和RaTG13在许多关键基因组特征上还是不同的,可以说,其中最重要的是SARS-CoV-2在S蛋白的S1亚基和S2亚基交界处含有一个多碱性切割位点(polybasic cleavage site)插入。尽管位于S1/S2切割位点附近的多碱性位点存在于包括HCoV-HKU1在内的其他人类冠状病毒和高致病性的禽流感病毒毒株中,但是这种可能增加了SARS-CoV-2传染性的插入并不存在于相关的蝙蝠β冠状病毒中。此外,SARS-CoV-2和RaTG13的受体结合结构域(RBD)仅有大约85%的相似度,并且在RBD的六个关键氨基酸残基中仅有一个是相同的。重要的是,序列和结构比较都提示着SARS-CoV-2 RBD非常适合与也被SARS-CoV使用的人ACE2受体结合。重要的是,最近在2019年中期从云南省另一种菊头蝠中取样的病毒(RmYN02)中观察到了在S1/S2切割位点上氨基酸PAA的独立插入,这表明这些插入事件反映了冠状病毒正在进行的进化的一个自然部分。虽然RmYN02在S蛋白上与SARS CoV-2相对不同(?72%序列相似度),但是在较长的复制酶基因上,它与这种人类冠状病毒具有最为密切的亲缘关系(?97%的核苷酸序列相似度)。
尽管SARS-CoV和MERS-CoV与SARS-CoV-2密切相关并且都有蝙蝠宿主,但这两种病毒之间的生物学差异却很明显。如上所述,SARS-CoV-2的传染性明显更高,从而导致与SARS-CoV和MERS-CoV完全不同的流行病学动态。在后两种病毒中,病例数的增长相对较慢,而MERS-CoV从未完全适应人类传播:大多数病例是由于阿拉伯半岛的骆驼溢出造成的,仅零星地发生人与人之间的传播。相比之下,SARS-CoV-2在当地的显著传播最让人吃惊。确定支撑这种传播能力的病毒学特征显然是当务之急。
SARS-CoV-2的人畜共患病起源
COVID-19的出现和迅速传播标志着一场完美的流行病风暴。这是一种毒性相对较高的呼吸道病原体,它来自一个具有跨越物种界限的不寻常本领的病毒家族。它出现在一个主要的人口中心和旅游中心,就在一年中最大的旅游旺季---中国春节---前夕。确实,流行病学模型表明SARS-CoV-2在武汉市被严格隔离之前已经在中国广泛传播也就不足为奇了。
早期基因组比较显示这种与SARS-CoV关系最密切的病毒也来自蝙蝠,这也不足为奇。近年来的采样已经确定了一系列令人印象深刻的蝙蝠冠状病毒,包括RaTG13和RmYN02。因此,对于多种冠状病毒而言,蝙蝠无疑是重要的宿主。尽管如此,蝙蝠在SARS-CoV-2的人畜共患病起源中所起的确切作用尚未确定。特别是,这些与SARS-CoV-2关系最密切的蝙蝠冠状病毒是从距武汉市1500多公里的云南省的动物中取样的。湖北省的蝙蝠冠状病毒相对较少,而且那些已被测序的蝙蝠冠状病毒在系统进化树上与SARS-CoV-2距离相对较远。基于此的一个简单的推断是,我们对蝙蝠病毒的采样强烈偏向某些地理位置。这将需要在以后的研究中加以纠正。此外,尽管96%~97%的序列相似度听起来像是现有的蝙蝠病毒与SARS-CoV-2密切相关,但是实际上这可能代表20多年的序列进化(然而,如果这种病毒在人类中有很强的适应性进化,那么潜在的分子时钟可能以不确定的速度滴答作响)。因此,几乎可以肯定的是,更多的采样将会鉴定出与SARS-CoV-2亲缘关系更近的其他蝙蝠病毒。一个关键问题是这些病毒或来自任何其他动物的病毒是否包含这些关键的RBD突变以及与SARS-CoV-2中相同的弗林蛋白酶样切割位点插入。
尽管蝙蝠很可能是这种病毒的天然宿主,但它们与人类之间的生态隔离使得其他哺乳动物物种很可能充当“中间”或“扩增”宿主,在这些中间宿主中,SARS-CoV-2能够获得在人类中有效传播所需的部分或全部突变。就SARS-CoV和MERS-CoV而言,果子狸和骆驼分别起着中间宿主的作用,尽管MERS-CoV可能在骆驼体内存在了几十年,然后才在人类身上出现,但在多次跨物种事件中,这些动物可能被更好地认为是真正的宿主。为了确定这些中间宿主物种可能是什么,必须对来自湿货市场或居住在人群附近的动物进行更广泛的采样。最近在非法进口到中国南部(广东和广西)的马来亚穿山甲中发现了与SARS-CoV-2密切相关的病毒,这突出说明了这一点。广东穿山甲冠状病毒在RBD上与SARS-CoV-2特别密切相关,包含认为使得与ACE2受体结合的六个关键突变中的全部六个(尽管它们在基因组的剩余部分上与SARS-CoV-2存在更大的差异)。尽管穿山甲因经常被人们非法贩运和处于濒危状态而引起人们极大的兴趣,但是穿山甲携带与SARS-CoV-2相关的病毒强烈表明,各种哺乳动物中都存在大量相关的β冠状病毒但尚未取样。 虽然我们过去在冠状病毒中的经验表明,动物宿主---天然宿主和中间宿主---的进化解释人类SARS-CoV-2出现的必要条件,但是不能排除这种病毒在2019年12月首次被发现之前在一段‘隐性’传播时间内在人类中获得了一些关键突变。特别是,这种病毒可能比预期的更早在人群中出现(也许甚至最早并不在武汉出现),但是由于无症状感染病例,具有轻度呼吸道症状的病例,甚至零星的肺炎病例,它在用于监视和病原体识别的标准系统中并未被观察到。在这段隐性传播期间,这种病毒可能逐渐获得了关键突变(可能包括RBD和弗林蛋白酶切割位点插入),从而使得它能够完全适应人类。直到发生了一系列肺炎病例,我们才能够通过常规监视系统检测到COVID-19。显然,对呼吸道感染的回顾性血清学或宏基因组学研究将有助于确定这种情况是否正确,尽管这样的早期病毒可能永远不会被检测到。
另一个受到广泛关注的问题是SARS-CoV-2是否是一种重组病毒,以及这种重组是否可能促进了它的出现。一个复杂的因素是sarbevirus病毒和更广泛的冠状病毒经历了广泛的重组,因此区分有助于病毒出现的重组与“背景”重组事件并非易事。重组可见于sarbevirus病毒基因组的多个位置(包括在S蛋白中)以及与SARS-CoV-2密切相关的蝙蝠病毒中。比如,有证据表明SARS-CoV-2、RaTG13和广东穿山甲冠状病毒之间会发生重组,而RmYN02的基因组类似地受到重组的影响。但是,很难确定重组事件的确切模式和基因组谱系,尤其是因为许多重组区域可能较小,并且随着我们对更多与SARS-CoV-2相关的病毒进行采样,这些重组区域可能会发生变化。为了解决这些问题,再次有必要对动物种群中的病毒多样性进行更广泛的采样。
正在进行的SARS-CoV-2基因组进化
随着COVID-19流行病的发展,更多的病毒基因组已被测序。正如基于它们最近的共同祖先所预期的那样,来自武汉的最早样本包含相对较少的遗传多样性。虽然这可以阻止详细的系统进化树和谱系地理学推断,但是它确实表明武汉的公共卫生部门在检测第一批肺炎聚集性病例方面做得非常出色。但是,这种看似最近的共同祖先并不排除存在一个在人类中隐性传播的疫情爆发前时期。
尽管积累遗传多样性意味着如今有可能检测到SARS-CoV-2序列的不同系统进化树簇,但仅通过基因组比较很难确定这种病毒在全球人群中传播时是否固定了表型上重要的突变,而且任何此类声明都需要仔细的实验验证。考虑到RNA病毒特征性的高突变率,很明显在这种病毒基因组中将出现更多的突变,这些突变将有助于我们追踪SARS-CoV-2的传播。
但是,随着流行病的蔓延,我们的序列样本量相对于病例总数来说可能非常小,以至于很难(如果不是不可能的话)检测每个传播链。因此,在尝试推断确切的传播事件时必须格外小心。顺便说一句,尽管冠状病毒的突变率可能比其他RNA病毒低,这是由于3'→5'核糖核酸外切酶内在地具有一定的校对活性,但它们的长期核苷酸替换速率(即分子进化)分布与其他RNA病毒相同。这提示着较低的突变率在一定程度上被宿主内较高的病毒复制速率所补偿。尽管没有证据表明这种突变能力(对RNA病毒常见)会导致表型---比如传播性和毒力---发生任何根本性变化,这是因为这些变化很少在单个疾病疫情规模上发生变化,但是监视病毒传播时表型的任何变化显然很重要。很有可能,COVID-19的病例数和/或病死率的任何下降都可能是由于人群免疫力的提高和流行病学背景而不是这种病毒中的突变变化引起的。
结论
SARS-CoV-2似乎将不可避免地成为人类中的第五种地方性冠状病毒(其他四种为HCoV-OC43、HCoV-229E、HCoV-NL63和HCoV-HKU1),并且目前正在完全易感人群中传播。冠状病毒显然具有跨越物种界限并适应新宿主的能力,这使得预测未来会有更多的病毒出现变得很简单,不过尚不清楚为何相比于一些其他的RNA病毒,冠状病毒具有这种能力。至关重要的是,对动物冠状病毒的监视应包括蝙蝠以外的动物,这是因为中间宿主的作用可能非常重要,从而为病毒在人类中的出现提供了更直接的途径。
鉴于野生动物中病毒的多样性以及它们的持续进化,可以说,降低未来疫情爆发风险的最简单最具成本效益的方法是尽可能限制我们对动物病原体的接触。尽管我们与动物世界的亲密关系意味着我们无法建立坚不可摧的屏障,但是针对非法野生动物贸易采取更强有力的行动,以及将所有野生哺乳动物(也许还有禽类)从湿货市场中清除将提供一个重要的缓冲。(生物谷 Bioon.com)
参考资料:
Yong-Zhen Zhang et al. A Genomic Perspective on The Origin and Emergence of SARS-CoV-2. Cell, 2020, doi: 10.1016/j.cell.2020.03.035.
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
87%用户都在用生物谷APP 随时阅读、评论、分享交流 请扫描二维码下载->
