
Nature:通过全球宏基因组分析,将已知的蛋白家族数量翻倍
来源:生物谷原创 2023-10-19 10:31
想象一下,科学家们用手电筒探索一个黑暗的房间,却只能清楚地辨认出光束范围内的东西。说到微生物群落,他们历来无法看到光束之外的东西---更糟糕的是,他们甚至不知道这个房间有多大。
想象一下,科学家们用手电筒探索一个黑暗的房间,却只能清楚地辨认出光束范围内的东西。说到微生物群落,他们历来无法看到光束之外的东西---更糟糕的是,他们甚至不知道这个房间有多大。
在一项新的研究中,来自美国劳伦斯伯克利国家实验室和加州大学伯克利分校等研究机构的研究人员通过研究微生物群落中的蛋白功能,以一种新颖的方法更好地了解微生物群落,从而凸显了微生物大量的功能多样性。相关研究结果于2023年10月11日在线发表在Nature期刊上,论文标题为“Unraveling the functional dark matter through global metagenomics”。

"论文第一作者 Georgios Pavlopoulos 现为亚历山大-弗莱明生物医学科学研究中心的研究主任。"这是用大规模并行计算对13亿个蛋白质进行的大规模分析"。
论文共同通讯作者Georgios Pavlopoulos说,“到目前为止,我们已将已知的蛋白家族数量增加了一倍以上,并确定了许多新的结构预测。这是对13亿个蛋白进行大规模并行计算的大规模分析。”
在美国能源部联合基因组研究所(JGI)的科学家们的指导下,这些作者开始了揭开隐藏在“黑暗”功能领域的神秘面纱的任务。他们的重点是破译错综复杂的蛋白功能多样性世界:尚未揭开面纱的微生物中的新蛋白家族和新功能。他们利用 26000 多个微生物组数据集的集体力量,成功编制了新型宏基因组蛋白家族(Novel Metagenome Protein Families, NMPF)目录。
论文共同通讯作者Nikos Kyrpides说,“我们如今可以通过与这些蛋白家族进行比较来分析新的数据集,或者进一步分析这些蛋白家族,以预测新的功能。”
揭示功能性“暗物质”
微生物群落无处不在,从土壤和胃部到深海,当涉及到能量循环时,它们能够做很多独特的事情---将生物量(biomass)转化为乙醇或氢,或将太阳能转化为氢。
微生物群落也非常难以研究。其中的许多微生物无法在实验室环境中培养。由于每个微生物群落都有自己独特的微生物组成和功能,因此不可能人为地复制整个群落。
宏基因组测序使得人们能够通过对样本进行全基因组测序来研究这些微生物群落中的完整基因构成,而无法区分哪个基因属于微生物群落中的每个微生物物种。因此,这一过程需要参考现有的基因组序列。
其中的一些蛋白被科学家们称为“已知的已知(known knowns)”,即它们与具有已知功能的基因相似。另一些则被称为“已知的未知(known unknowns)”,也就是说,它们与以前从分离出的有机体中获得的已知基因相似,但我们仍不确定它们的功能。
然而,如果微生物群落中的某个基因与以前从分离出的有机体中获得的已知基因不匹配,科学家们就无法得知其功能或来源。因此,这些基因通常被当作无用信息而从任何分析中剔除。这些基因代表着“未知的未知(unknown unknowns)”,因为它们与我们已经确定的任何基因都不相似。
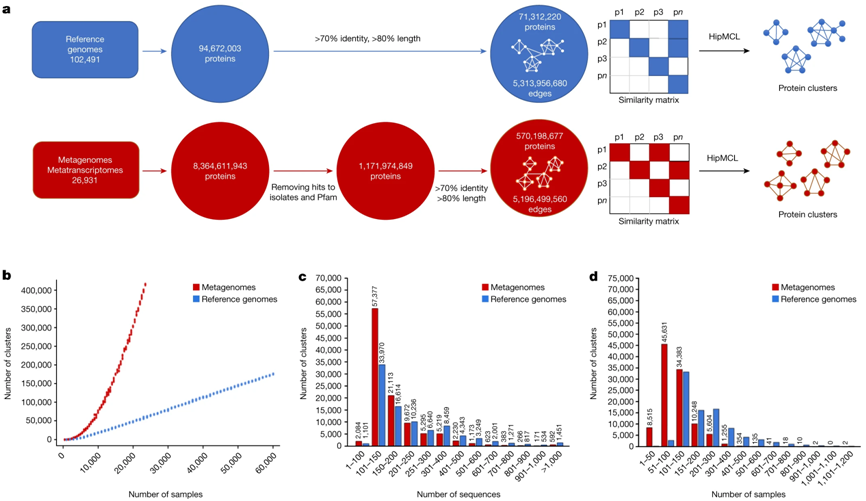
序列聚类分析概述。图片来自Nature, 2023, doi:10.1038/s41586-023-06583-7。
Kyrpides说,“在我们目前已知的蛋白家族中,有很大一部分---大约30%~50%---仍然没有任何已知功能,但我们知道这些蛋白家族。然而,将近20年的宏基因组数据和宏基因组分析,仍然没有对来自宏基因组本身的蛋白家族进行真正的分析。”
最近,其他的研究团队利用人工智能的力量解码了蛋白序列的语言,并获得了关于它们的可能功能的提示。然而,这些研究工作仅限于已知的蛋白序列领域。
Pavlopoulos说,“在这项的研究中,我们不仅涉足了未知领域来了解功能多样性的广阔前景,而且还通过应用人工智能方法来揭示它们的作用,从而突破了界限。因此,我们积累了大量开创性的见解,极大地拓展了各类蛋白潜在功能的视野,包括那些在生物技术领域有着关键应用的蛋白,如DNA编辑酶。”
以全新方式利用蛋白家族
近年来,新蛋白家族的发现开始趋于平稳,这或许表明科学家们已经“捕捉”到了大量的多样性,尽管还没有确定它们的具体功能。但是,这些“未知的未知”可能蕴藏着怎样的多样性呢?
这些作者从来自IMG (Integrated Microbial Genomes & Microbiomes)数据库的80亿个宏基因组基因开始(还参考了来自JGI的地球微生物组基因组数据)。然后,他们剔除了任何与之前已知基因有一点相似的基因,剩下大约 12 亿个新基因。他们将剩下的这些基因聚类成族。在此基础上,他们将重点放在至少有 100 个成员的家族上。
Kyrpides解释说,“如果你有100个序列,那么这种聚类分析的质量就会大大提高,因为很难有100个来自不同地点或栖息地的序列能够很好地随机对齐。重现100 次几乎是不可能的。”
当这些作者完成这一阶段的研究工作后,他们发现在这个宏基因组空间(“未知的未知”)中,蛋白家族的多样性远远超过参考基因组,至少是参考基因组的两倍。
Kyrpides说,“随着我们不断增加样本,我们得到了更多的蛋白家族。再过几年,随着我们继续对更多的宏基因组进行测序,一些目前只有50个或更多成员的家族也将增加到100个或更多成员。因此,我们说多样性增加了一倍,但实际上可能会增加三倍、四倍、五倍或十倍。”
进一步挖掘多样性
虽然这些作者没有深入研究功能,但是他们能够进一步描述这些家族的特征。他们按照环境将这些蛋白家族进行分类,发现只有7%的蛋白家族在所有八个环境类别中共享。相反,蛋白家族偏爱特定的环境---无论是土壤、动物宿主还是海洋生态系统等。
Pavlopoulos解释说,“因此,它们一定是在为那个栖息地做一些有趣或重要的事情。这绝对是科学界如今可以进一步利用的材料。比方说,有人正在研究土壤环境或人体---他们可能会选择其中的一些蛋白科学,并尝试从功能上描述它们的特征,因为它们对该栖息地来说非常特殊。”
分类分析发现这些蛋白家族大部分属于细菌和病毒,不过有600万个序列无法加以分类。这些作者还试图通过三维建模来确定这些基因的功能,并将未知基因的结构与已知基因的结构进行比较---相似的结构意味着相似功能的可能性很高。他们还发现了具有全新结构的蛋白家族。
这是首次利用蛋白结构来帮助描述大量微生物暗物质的特征。这项研究耗时约两年完成,当时只测序了约2万个宏基因组。如今,这个数字接近 6 万。
Kyrpides说,“在已知的微生物多样性中,仍有70%~80%的基因组尚未被捕获。因此,在功能多样性方面,这种多样性肯定也蕴藏着许多新的秘密。”(生物谷 Bioon.com)
参考资料:
1. Georgios A. Pavlopoulos et al. Unraveling the functional dark matter through global metagenomics. Nature, 2023, doi:10.1038/s41586-023-06583-7.
2. Doubling Down on Known Protein Families
https://newscenter.lbl.gov/2023/10/11/doubling-down-on-known-protein-families/
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
