
MBD2作为抑制因子通过调节STAT1-IFN-γ轴来维持1型糖尿病Th1程序的稳态
来源:本站原创 2021-08-30 17:24
甲基-CpG结合结构域2 (MBD2)通过与甲基化的CpG DNA结合来解释DNA甲基组编码的信息,并在转录水平上调控靶基因的表达。
甲基-CpG结合结构域2 (MBD2)通过与甲基化的CpG DNA结合来解释DNA甲基组编码的信息,并在转录水平上调控靶基因的表达。虽然DNA甲基化脱位长期以来被认为可以触发或促进1型糖尿病(T1D)的自身免疫反应,但MBD2在T1D发病机制中的确切作用尚不明确。在这里,作者在NOD背景下构建了Mbd2敲除模型,发现Mbd2缺陷加剧了NOD小鼠自发T1D的发展。总的来说,作者的数据表明MBD2可能是一个可行的目标,在临床环境中开发基于表观遗传学的T1D治疗方法。

图片来源:https://www.nature.com/articles/s41418-021-00852-6
1型糖尿病(T1D)是一种由T细胞介导的胰岛β细胞破坏引起的慢性自身免疫性疾病。因此,与外源性胰岛素治疗相比,旨在抑制自身免疫反应或诱导自我耐受的策略在促进β细胞恢复和预防疾病进展方面有更好的前景。研究认为,CD4效应T细胞通过产生多种促炎细胞因子,协调细胞毒性CD8 T细胞、B细胞和巨噬细胞的功能,在免疫失调中发挥核心作用,从而引发严重的胰岛素炎症并导致β细胞破坏。在CD4效应T细胞的不同亚群中,Th1和Th17是促成T1D发病的两个关键成分。Th1细胞是I型炎症细胞因子的主要来源,尤其是IFN-γ、TNF-α和IL-1β,它们能诱导β细胞凋亡。相比之下,Th17细胞具有更强的致病性,事实上,糖尿病致敏细胞的特征通常是将非致病性的Th17细胞转化为致病性的Th17细胞,同时赋予th1样特性。因此,Th1细胞被认为是T1D发病机制中最关键的致病亚群。
虽然遗传易感性使患者具有较高的T1D风险,但与环境线索相关的表观遗传因素也在触发对胰腺β细胞的自身免疫反应中发挥了关键作用。特别是DNA甲基化,与基因转录调控相关的重要表观遗传机制之一,已经被认为参与了T1D发病机制。例如,对T1D不一致单合子双胞胎的研究发现了130多个不同的甲基化CpG位点,其中一些位于T1D易感区域,如HLA类II、TNF和GAD2。DNA甲基化编码的信息由甲基-CpG结合域(MBD)蛋白家族解释,该蛋白家族包含11个已知成员,但只有Mecp2、MBD1、MBD2和MBD4通过直接与甲基化的CpG
DNA结合来调控基因转录。在与甲基化的CpG DNA结合后,它们形成一个抑制复合物或与核小体重塑和去乙酰化酶(NuRD)复合物串扰,以实现一个连贯的转录程序。NuRD通过与不同的组蛋白去乙酰化酶和染色质重塑因子(如组蛋白去乙酰化酶HDAC1/2、atp依赖的重塑酶CHD3/4、组蛋白伴侣RBBP4/7等)合作发挥多种功能。NuRD复合物的纯化和分析表明,MBD2和MBD3可以形成相互排斥的NuRD复合物,每一个都以不同的亲和力和选择性识别甲基化的DNA。MBD2基因敲除小鼠是存活的,而MBD3基因敲除小鼠是胚胎致命的。重要的是,MBD2对甲基化模式具有更高的选择性,并已被确认在免疫反应的调节中。
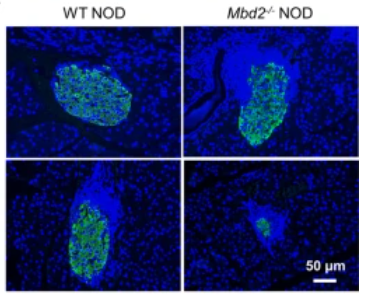
在NOD小鼠中,MBD2缺乏会加重T1D的发展
图片来源:https://www.nature.com/articles/s41418-021-00852-6
此前,作者证明了MBD2在C57/B6小鼠Th17分化和实验性自身免疫性脑脊髓炎(EAE)发展中不可或缺的作用。在这份报告中,作者发现Mbd2缺乏会加重NOD小鼠的T1D。Mbd2 / CD4 T细胞过继转移到NOD。scid小鼠还通过促进Th1极化增强T1D发病。在机制上,MBD2选择性地结合Stat1启动子中的甲基化CpG DNA,通过抑制Stat1 - ifn -γ信号来维持Th1程序的稳态,在T1D患者中也获得了类似的结果。总的来说,作者的发现揭示了MBD2在T1D发病机制中的作用,这可能为在临床环境中开发基于表观遗传的T1D治疗铺平道路。(生物谷 Bioon.com)
参考文献
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
