
J Adv Res:IFP35通过促进nrf2调控的铁死亡加重金黄色葡萄球菌感染
来源:生物谷原创 2024-07-26 10:11
本研究提示IFP35对SA感染引起的免疫病理有不良影响,铁死亡参与了sa诱导的病理过程,IFP35的缺失减少了sa介导的铁死亡。
金黄色葡萄球菌(SA)是一种革兰氏阳性球菌,可引起危及生命的疾病,如肺炎和其他感染。SA对肺的侵袭是急性肺损伤(ALI)和急性呼吸窘迫综合征的重要诱因。虽然抗生素可以减轻SA感染,但新的耐药菌株的出现大大增加了与这些感染相关的死亡率。因此,除了开发新的抗生素外,迫切需要替代和/或辅助治疗。
干扰素诱导蛋白35 (IFP35)家族蛋白,包括N-myc和stat相互作用蛋白(NMI),在抗病毒应答中发挥重要作用。lps诱导的脓毒症休克小鼠模型中,IFP35缺失减轻了炎症反应,提高了存活率。虽然IFP35在先天免疫中的功能是众所周知的,但其在细菌感染,特别是SA感染中的病理作用和分子调控机制仍然知之甚少。
铁死亡是一种新的程序性细胞死亡形式,由过量的游离铁和脂质过氧化物介导。铁死亡与细菌感染密切相关。结核分枝杆菌可诱导巨噬细胞铁死亡,促进细菌扩散,加重组织坏死铜绿假单胞菌通过分泌脂氧合酶氧化花生四烯酸磷脂酰乙醇胺诱导人支气管上皮细胞铁死亡。SA诱导囊性纤维化小鼠模型肺脂质过氧化。然而,sa感染的细胞是否会发生铁死亡尚不清楚。
核因子红细胞2相关因子2 (Nrf2)是一种转录因子,在细胞防御氧化应激中起着至关重要的作用。多项研究表明Nrf2可以通过多种方式调节铁死亡。此外,Nrf2通过下调参与铁摄取的转铁蛋白受体1和二价金属转运蛋白1的表达来减少铁摄取。Nrf2激活还通过上调谷氨酸-半胱氨酸连接酶(GSH生物合成中的限速酶)的表达来增强谷胱甘肽合成(GSH)。此外,Nrf2还可缓解铁中毒相关的ALI和糖尿病肾病。尽管Nrf2在嗜铁细胞凋亡介导的病理生理中的作用已被广泛证实,但其在介导的免疫病理中的作用以及调控Nrf2的可能机制在很大程度上仍然未知。

图片来源:https://doi.org/10.1016/j.jare.2023.09.042
近日,来自浙江大学医学院的研究者们在J Adv Res杂志上发表了题为“IFP35 aggravates Staphylococcus aureus infection by promoting Nrf2-regulated ferroptosis”的文章,该研究表明IFP35通过促进Nrf2的泛素化和降解来促进铁凋亡,从而加剧SA感染,靶向IFP35可能是治疗SA引起的感染性疾病的一种有前景的方法。
严重金黄色葡萄球菌(SA)感染是危及生命的疾病之一。干扰素诱导蛋白35 (IFP35)是一种参与多种生物学功能的多效因子,但其在SA感染中的生物学作用尚不完全清楚。铁死亡是一种由游离铁和有毒脂质过氧化物的增加驱动的新型受调控的细胞死亡,在组织损伤中起重要作用。铁死亡是否参与sa诱导的免疫病理及其调控机制尚不清楚。本研究者旨在确定IFP35在sa诱导的肺部感染中的作用和潜在机制。
研究者采用野生型(WT)和IFP35敲除鼠或巨噬细胞建立SA感染模型,进行组织学分析以评估肺损伤。采用实时荧光定量PCR、western blotting、流式细胞术、共聚焦显微镜检测铁死亡,利用Co-IP和免疫荧光技术阐明了分子调控机制。
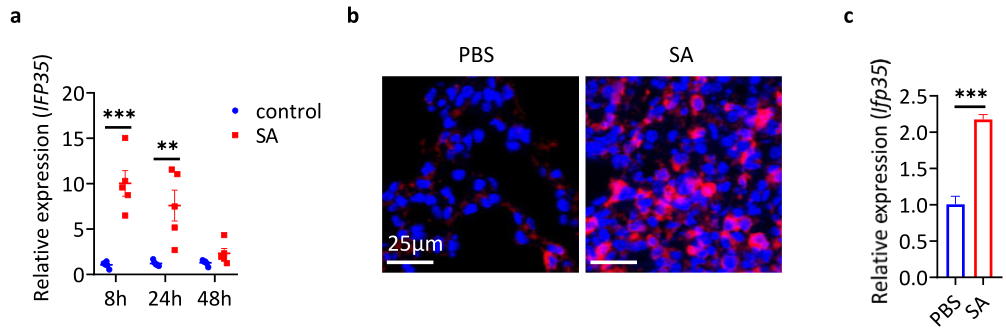
缺乏IFP35可减轻金黄色葡萄球菌感染后肺损伤
图片来源:https://doi.org/10.1016/j.jare.2023.09.042
研究者发现sa感染小鼠的巨噬细胞和肺组织中IFP35水平升高。IFP35缺乏对sa诱导的小鼠肺损伤有保护作用。此外,SA感染后发生铁死亡并导致肺损伤,IFP35缺乏可改善肺损伤。在机械上,IFP35促进了核因子e2相关因子2 (Nrf2)的泛素化和降解,加重了sa诱导的铁下沉和肺损伤。
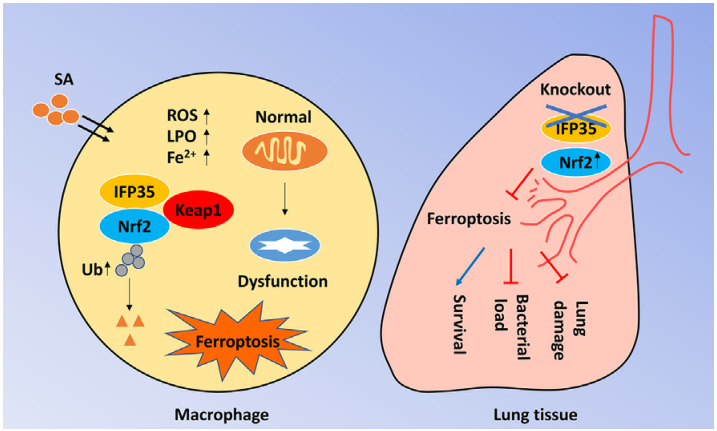
IFP35通过促进nrf2调控的铁死亡加重金黄色葡萄球菌感染
图片来源:https://doi.org/10.1016/j.jare.2023.09.042
综上所述,本研究提示IFP35对SA感染引起的免疫病理有不良影响,铁死亡参与了sa诱导的病理过程,IFP35的缺失减少了sa介导的铁死亡。机制研究表明,IFP35缺乏通过抑制Nrf2的泛素化和降解来上调Nrf2,从而减少SA诱导的铁凋亡。本研究为SA感染的发病机制提供了新的见解。特异性靶向IFP35或铁死亡是治疗SA相关疾病的一种很有前景的策略。(生物谷 Bioon.com)
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
