
Briefings in Bioinformatics:南方科技大学医学院杨亮和环境学院夏雨科研团队开发一种快速灵敏诊断病原细菌及其携带耐药基因的宏基因组学工作流程
来源:南方科技大学医学院 2022-10-20 13:09
本文报道了一个对临床样本中的病原细菌及其携带耐药基因进行快速诊断的宏基因组学工作流程。
2022年10月18日,南方科技大学医学院杨亮和环境学院夏雨科研团队联合在Briefings in Bioinformatics杂志上在线发表了题为“A rapid bacterial pathogen and antimicrobial resistance diagnosis workflow using Oxford Nanopore adaptive sampling sequencing method”的研究论文。本文报道了一个对临床样本中的病原细菌及其携带耐药基因进行快速诊断的宏基因组学工作流程,该流程整合了基于GPU运算的Nanopore适应性采样“人类DNA过滤”测序模式和自主研发的实时物种鉴定/抗性基因预测分析工具RUARGpore(https://github.com/sustc-xylab/RUARGpore.git),实现了上机4.5小时内病原及所携带耐药基因的精准检测。同时,该方法相比现有宏基因组学诊断方法有效的降低了成本,有利于大规模临床推广。
l 背景介绍及研究概述
高通量测序已成为检测和鉴定环境样本和临床样品中病原微生物的有效技术平台,极大增强了诊断和跟踪传染病病原的能力,尤其是在诊断未知突发性病原体时相对于传统培养学方法及分子诊断具有显著的优势。然而,临床样本的复杂性对宏基因组测序诊断提出了艰巨的挑战。由于病原体负荷的变化、采集部位共生菌群的存在以及宿主细胞的高占比,宏基因组测序分析必须处理临床样本中的少量原核DNA和大量宿主DNA,这大大降低了微生物检测的整体分辨率,极大浪费测序成本,延长测序时间,影响后续病原体序列的检出和耐药基因的鉴定。纳米孔适应性采样(nanopore adaptive sampling,NAS)测序直接在测序过程中通过快速的GPU运算实现对目标序列(例如人类DNA)的实时比对,以确定经过纳米孔的DNA分子是否应该进一步测序(接受)或直接从孔中移除(拒绝)。本研究开发了一个对病原细菌和抗生素耐药基因(ARG)进行快速精准诊断的宏基因组学工作流程,该流程整合了基于NAS测序的“人类DNA过滤”模式和实时物种/抗性基因预测的分析工具RUARGpore,能实现临床样本DNA上机后自动化分析诊断。研究团队使用该流程对21个支气管肺泡灌洗液(BALF)样本进行了分析,将微生物序列富集至少8倍,并在4.5小时内从物种水平上准确检测到ARG。
l 研究内容
1. 建立快速精准诊断病原细菌及其携带耐药基因的宏基因组学工作流程
本研究开发了一种新的mNGS工作流程,将NAS的实时测序数据流与分析工具RUARGpore相结合,快速精准的检测病原体和ARG,缩短检测周转时间。工作流程包括临床样本DNA提取、文库制备、GridION NAS测序和RUARGpore软件分析。该工作流程通过“人类DNA过滤”增加了临床样本中微生物序列的整体测序深度,而测序过程本身不改变微生物组成,从而在4.5 h内完成复杂临床样本中低丰度病原体的鉴定和ARG检测。
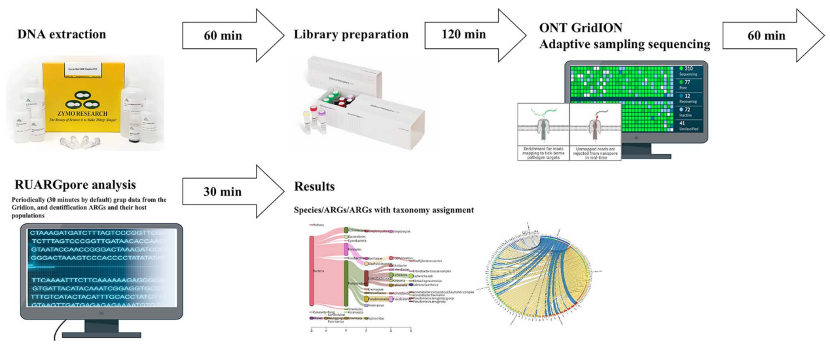
图1 宏基因组管道的示意图。mNGS工作流程包括临床样本DNA提取、文库制备、GridION NAS测序和ARGpore2软件分析。样本采集后最多需要3小时即可完成DNA 提取和纳米孔文库制备,并且可以在启动NAS测序运行后1小时内使用AMR基因分型进行物种鉴定,平均周转时间为4.5小时
2. 该检测流程对低丰度微生物的富集效果
分析NAS测序结果,从模拟样品中富集的微生物序列是正常测序方法中检测到微生物序列的2.7倍。NAS测序结果同时提供了足够的测序深度(即>>×基因组覆盖率),可以较好的组装模拟群落中的七种细菌。使用NAS测序从4922条与耐药基因库匹配的序列中识别出124个ARGs,其中119个ARGs是定位在7个细菌基因组上。以tet基因家族介导的四环素抗性为例,使用NAS测序再开始测序的5min内就检测到两条比对到tet基因的序列。
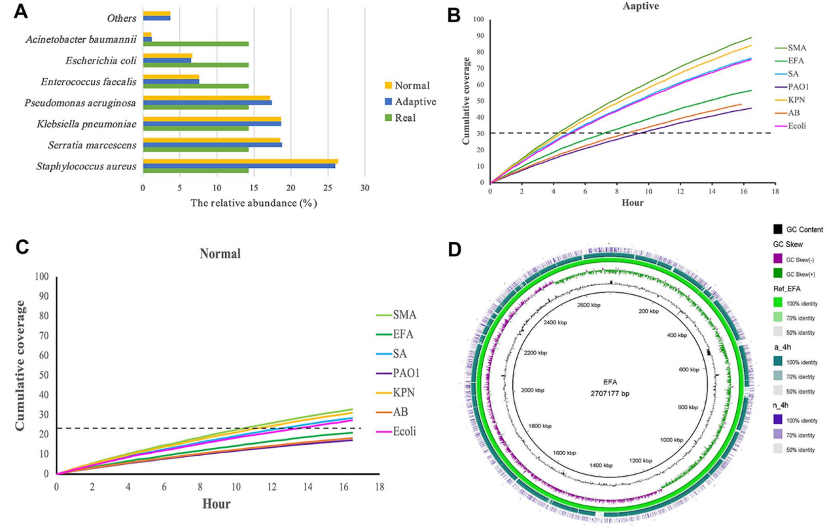
图2 用NAS测序富集模拟群落中的微生物物种。a) NAS 和对照运行中七种微生物物种丰度的条形图。 NAS 方法清楚地显示了7种微生物与对照的比例非常相似,但都与模拟样品中的真实比例有显着差异。b)和 c)7种微生物 NAS(b)和正常测序(c)的物种基因组覆盖率。d) 4 h测序后粪肠球菌基因组组装。
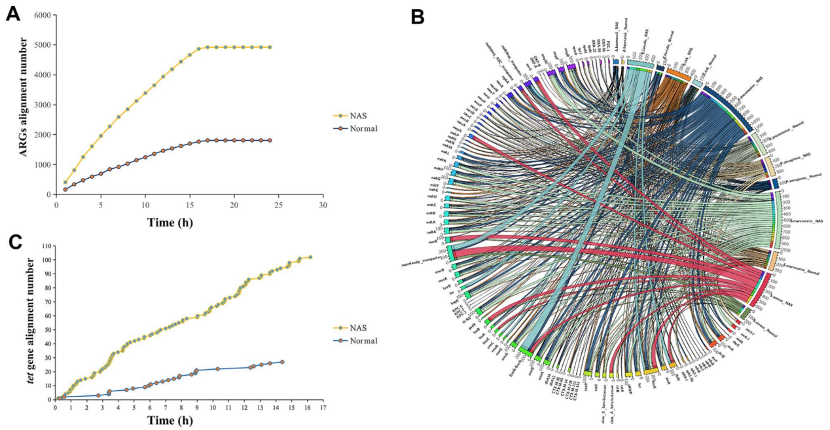
图3 使用NAS与正常测序检测ARGs。a) NAS与正常测序的ARGs比对。 b) 弦图说明ARG与7种携带ARG的致病物种之间的相关性。c) NAS与正常测序的tet基因比对数。正常测序的前5分钟没有检测到tet基因,而在同一时间点在NAS测序中检测到两个tet基因。
3. 使用该检测流程与常规Nanopore测序流程在同一张芯片上检测BALF 1样品的效果对比
将两种海洋古菌(S. acidocaldarius:H. mediterranei=4:1)纳入 B1 样本作为内部参考。S. acidocaldarius与H. mediterranei的比率在NAS测序中为3.6:1(读数 498:137),在ONT正常测序中为2.6:1(读数 132:51),而在 Illumina测序中未检测到H. mediterranei。常规微生物学培养检测到的病原微生物(肺炎克雷伯菌和鲍曼不动杆菌)用三种测序方法都检测到。但不同的是,鲍曼不动杆菌在三种方法中检测到的丰度最高10个物种占有一席之地,但肺炎克雷伯菌仅在NAS测序和ONT常规测序方法中排名前十。NAS 测序运行的第1小时中检测匹配为肺炎克雷伯菌和鲍曼不动杆菌的序列,分别是ONT常规测序方法检测到的190倍和16倍。使用NAS测序方法,发现肺炎克雷伯菌携带属于15种AMR类型的63个ARGs,鲍曼不动杆菌携带属于7种AMR类型的31个ARG。使用正常的ONT测序方法,未检测到病原体携带的ARGs。在NAS序列运行的第 1h,检测到42个ARGs(属于11种不同的AMR类型);6小时,检测到鲍曼不动杆菌和肺炎克雷伯菌分别携带的7种AMR类型(28个ARG)和15种AMR类型(56个ARG)。
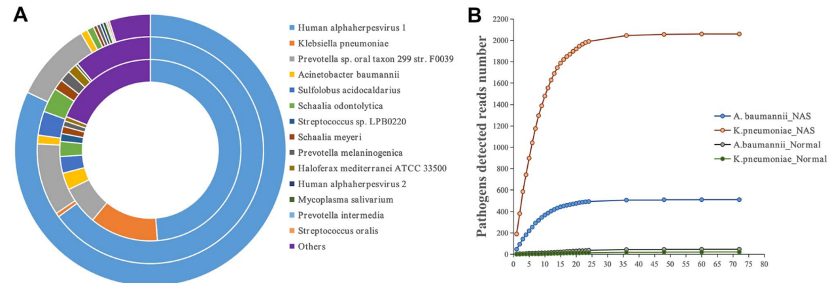
图4 在B1样本中使用NAS与正常测序进行物种检测。a) NAS、ONT 正常测序和 Illumina 测序检测到的前十种物种的丰度(内环为 NAS,中央为 ONT 正常测序,外环为 Illumina 测序)。b) NAS与正常测序的病原体(肺炎克雷伯菌和鲍曼不动杆菌)比对数。
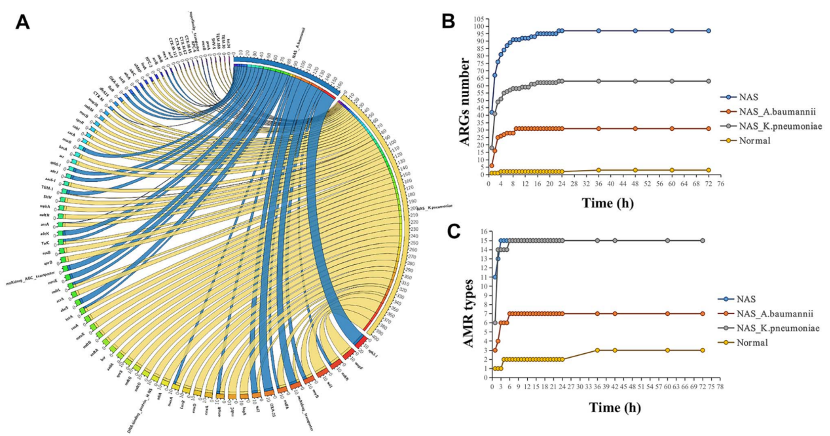
图5 NAS和正常测序检测B1样本中的ARGs。a) 弦图说明ARGs与2种携带ARG的致病物种之间的相关性。b) NAS与正常测序的ARGs比对数。c) AMR类型检测到的NAS数量与正常测序的比较。在NAS序列运行的6小时中,检测到鲍曼不动杆菌和肺炎克雷伯菌分别携带7种AMR类型(28个ARGs)和15种AMR 类型(56个ARGs)。
4. 应用该流程使用单个ONT测序芯片同时对五个BALF样品进行检测的效果展示
Shannon alpha 多样性分析表明,Illumina 平台测序的样本始终比 NAS 测序的样本具有更高的多样性。使用 NAS 测序方法在常规微生物学阳性报告样本中检测到一致的病原微生物,在常规微生物学阴性报告样本中未检测到病原菌。该流程和实验室培养之间的物种水平PPA为100%(16/16),NPA为100%。除了在三个样本中检测到较少的肺炎链球菌(B2,B6,B7均 < 1000 reads)外,使用 NAS 测序方法在其他受实样本中检测到高丰度的病原体。在B2、B6和B7中NAS序列运行的前1小时,检测到的用于产生可靠病原体鉴定的测序读取数分别为16、24和13。相比之下,正常 ONT 测序的前 1 小时无法检测到这些病原体。
使用本流程生成的测序数据产生可靠微生物物种和 AMR 鉴定所需的最短测序运行时间。细菌物种的鉴定在所有时间点都是一致的,在NAS测序运行的1小时内宣布样品为微生物阳性所需的最低检测阈值。相比之下,检测到的 ARG 和预测的AMR表型的数量在测序运行过程中增加,大多数 AMR 表型是在测序运行的前6小时内预测的。总体而言,结果表明微生物鉴定可以在测序运行的第1小时内确定,而大多数 BALF ARGs可以使用6小时的测序运行来表征。
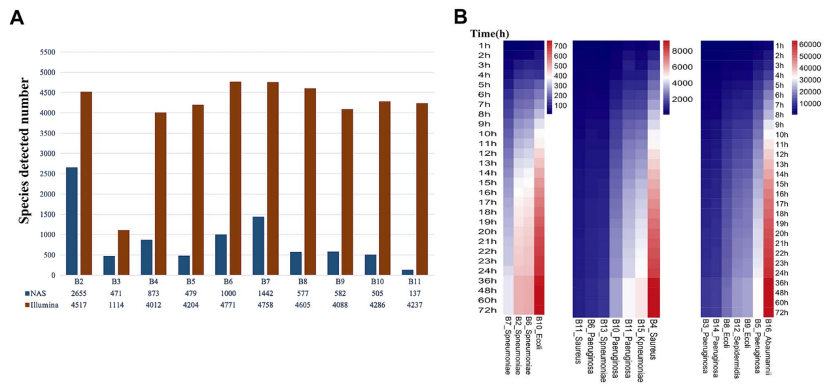
图6 在B2-B21样本中使用NAS测序进行物种检测。a) 通过NAS和Illumina测序检测到的物种数量。Shannon alpha多样性分析表明,Illumina平台测序的样本始终比 NAS测序的样本具有更高的多样性。b) NAS产生序列比对到病原体的数目。
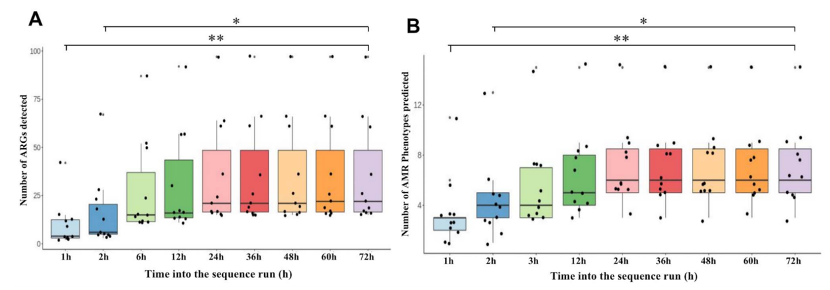
图7 在72小时的测序程序运行过程中检测ARGs。a) 和 b) 确定检测ARG和AMR表型所需的最佳测序时间。绘制每个样品中检测到的ARGs (a)和AMR表型(b)的总数,并使用Kruskal-Wallis H检验确定在不同时间点检测到的ARGs和AMR表型的统计显着性。实线,中位数;*,P值 < 0.05;**,P值 < 0.005。
l 总结展望
基于培养的微生物诊断和药敏试验已经使用了70多年,作为急性感染的临床管理指南存在一些局限性,主要是因为从样本到结果的传统检测时间周转缓慢。快速、准确的诊断将能够及时使用适当的抗生素进行治疗,并改善治疗结果和抗菌药物管理。我们开发了一种新的 mNGS 工作流程,在 4.5 h 内完成低丰度微生物标本中的病原体的鉴定和ARGs检测。这种时间差异在临床上具有重要意义,尤其是在快速鉴定ARG和检测厌氧菌和真菌方面。快速结果有可能限制不适当的经验性抗菌药物覆盖率,降低相关的发病率和死亡率。
本研究mNGS工作流程,可以在一张芯片上同时检测5例样本,成本约为每个样本267美元。通过批量购买流动池(R9),使用快速条形码测序工具(SQK-RBK004),可以同时对多个样本进行测序,从而进一步降低成本。相比之下,对BALF样本进行Illumina元基因组测序分析的成本为445美元,两者成本差异为178美元,这将大大减轻患者的经济负担。本文介绍的mNGS检测流程已经申请中国发明专利保护。
l 作者信息
南方科技大学医学院杨亮、南方科技大学环境学院夏雨为本论文共同通讯作者。南方科技大学医学院博士生程航和南方科技大学环境学院孙瑜鸿为本文第一作者。华中科技大学协和深圳医院邓名贵,余治健,南方科技大学第二附属医院(深圳市第三人民医院)朱刚,曲久鑫,刘磊等为该工作提供了重要临床应用指导。
l 通讯作者简介:

共同通讯作者 杨亮 教授/博士生导师
作者单位:南方科技大学医学院
作者简介:南方科技大学医学院教授,博士生导师,2004年本科毕业于南开大学生物科学专业,2009年在丹麦技术大学获得博士学位,随后在丹麦技术大学和德国汉堡大学从事博士后工作,2012年入职新加坡南洋理工大学生命科学学院做助理教授,在2018年获得终身教职 (副教授)和担任生命科学学院助理院长,同时兼任新加坡国家级卓越研究中心"新加坡环境生物工程中心(SCELSE)"公共卫生中心实验室副主任。杨亮于2018年底全职加入南方科技大学医学院任教授,是广东省杰出青年基金获得者,深圳市海外引进高层次人才,教育部青年特聘专家,新加坡南洋理工大学访问教授。在国际学术期刊包括Lancet, Nature Communications,Nature Protocols, Proc Natl Acad Sci U S A, Science Signaling, Trends in Microbiology, Nano Letters,Current Opinion in Biotechnology,Briefings in Bioinformatics等发表论文180多篇,论文被引用超过11000次,H-Index 52(Google Scholar),入选2019、2020 爱思唯尔中国高被引学者和2019、2020全球前2%顶尖科学家榜单。

通讯作者 夏雨 助理教授(副研究员)/博士生导师
作者单位:南方科技大学环境科学与工程学院
作者简介:夏雨博士,香港大学,水资源与环境工程博士。现任南方科技大学环境科学与工程学院副研究员,博士生导师。环境微生物与生态基因组学实验室负责人。研究兴趣集中于:结合BONCAT, Single-cell sorting等先进分子生物学手段、以Nanopore测序为代表的单分子高通量测序技术以及生物信息学大数据分析,解密生物处理反应器、极端自然系统及洁净室内环境中微生物群系的群落构建原理、功能调控机制以及关键基因(耐药基因)转移规律。近五年来在The ISME Journal, Environmental Science & Technology, Water Research 等顶级期刊发表论文40余篇,总引用次数 2600余次(Google Scholar),作为第一发明人申请发明专利3项。现任中国工程院院刊Engineering (SCI impact factor 12.834) 、Frontiers in Energy Research (SCI impact factor 3.858)以及iMeta 编委。应邀在国际会议做报告20余次,1次担任分会主席,1次大会报告;曾担任南方科技大学教授委员会环境科学与工程学院代表委员,美国微生物协会香港地区青年大使。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
