
纳米孔PromethION测序+Shasta完成人类基因组端粒到着丝粒的高效从头组装
来源:NanoporeTechnologies 2020-11-19 12:28
Shasta是一个用于纳米孔测序数据的从头组装和矫正算法,由加州大学圣克鲁兹分校(UCSC)和陈-扎克伯格倡议计划(CZI)联合开发。团队在2019年利用初代Shasta分析流程对纳米孔测序数据进行从头组装、矫正和Hi-C拼接Scaffold(图1),使用一台纳米孔PromethION测序设备在9天内完成了11个人类基因组测序,研究成果发表在《N
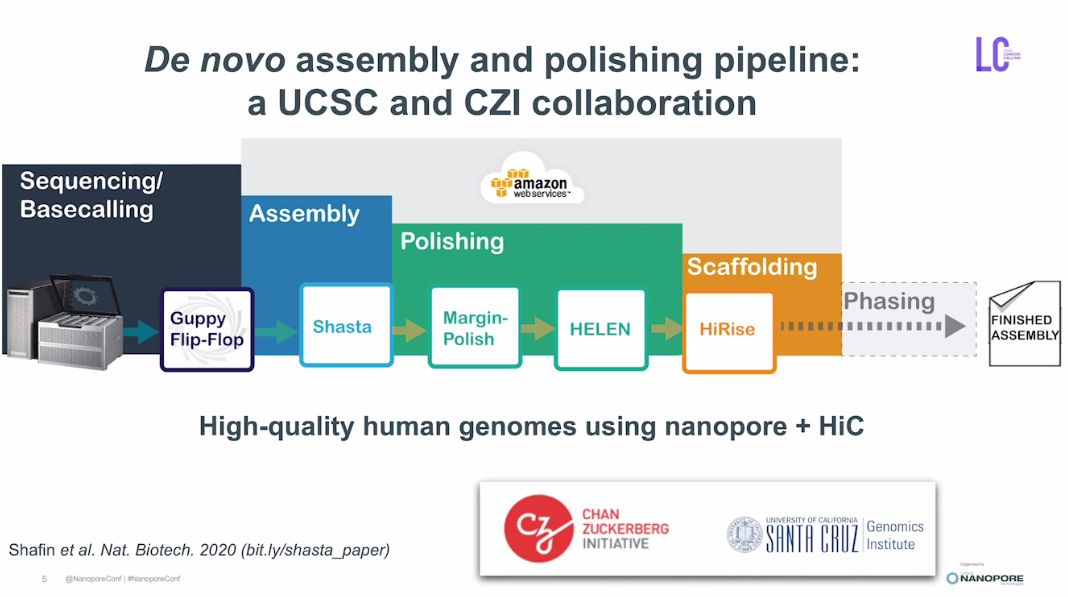
Shasta是一个用于纳米孔测序数据的从头组装和矫正算法,由加州大学圣克鲁兹分校(UCSC)和陈-扎克伯格倡议计划(CZI)联合开发。团队在2019年利用初代Shasta分析流程对纳米孔测序数据进行从头组装、矫正和Hi-C拼接Scaffold(图1),使用一台纳米孔PromethION测序设备在9天内完成了11个人类基因组测序,研究成果发表在《Nature Biotechnology》, DOI:
高通量数据:共生成2.4 Tb序列数据,10kb及以上读长序列的覆盖度中位数约为55X,100 kb及以上的超长读长序列覆盖深度中位数约为6.5X,N50介于20 Mb-30Mb之间。
成功组装困难区域:纳米孔长读长序列成功组装了此前难以组装的重复主要组织相容性复合体(MHC)区域,一条单一连续的Contig覆盖到了整个区域,获得Scaffold达整条染色体臂,甚至完整的染色体级。
降低时间及成本:初代Shasta算法组装一个基因组最快仅用不到6小时,每个样本成本约70美元,大幅降低时间周期和成本。
近期,Shasta更新到v0.4版,显着改进计算重叠的算法,引入了贝叶斯模型(Bayesian Model)预测均聚物的数量,新版本带来了巨大的算法优化:
进一步大幅提升连续性:NG50 提升50%,连续性升翻倍
使用最新版的纳米孔软件Guppy对原始数据重新进行识别碱基,团队利用不同人类样本对新版Shasta进行了性能测试。结果显示在标准人类基因组HG002中,NG50从约20Mb提升至约30 Mb,升幅约50%(图2)。在组装序列长度大于100 kb的超长读长数据集时,获得NG50约为58Mb,连续性几乎翻倍。在人细胞系CHM13样本上进行单倍体人类组装,获得NG50约为65Mb。
组装连续的染色体臂:
在人类参考基因组GRCh38版的组装中,其中一条Contig(8号)几乎跨越了11号的整个染色体臂(图3)。在标准基因组HG002样本中,仅使用来自3张测序芯片的数据,可以跨越大约7条染色体臂(标准Guppy 3.6组装),而超长序列组装出了近半的21条染色体臂,R10.3版测序芯片数据组装出了11条染色体臂。其中,预期的组装缺口是由于参考基因组和样本间的结构变异差异引起。
组装结果与其他“金标准”一致:
将Shasta组装与来自T2T联盟的人CHM13端粒到端粒组装结果(超链接:【精彩回顾】London Calling 2019——Day 2 研究精彩集锦)比较,显示23个染色体臂都有候选全长组装。聚焦在12号染色体这个例子上,显示在长臂(q)和短臂(P)的超级scaffold比对一致性超过了99.7%(图4)。
组装时间减半,组装完整性提升:Shastav4.0版+ 纳米孔软件Guppy最新版
从HG002基因组整体数据来看,Shasta结合最新版纳米孔软件Guppy可将组装时间从最初论文中描述的约6小时减少至约3小时;对超长序列数据集组装时间也从约15小时减少至不到6小时。基因组总长度也有所增加,插入缺失数量减少了约5倍,这都是用Guppy 3.6.0产生的结果(用R10.3组装数据甚至更佳)。基因组组装完整性(BUSCO)同样有大幅提升,几乎与GRCh38一致。
双倍型分型——联合基因分型与定向
“整条染色体上,长读长双倍型分型的表现优于短读长基因分型。在可定位性低的区域,由于短读长数据无法很好地定位,纳米孔长读长测序优势显着。”——Benedict Paten
通过与谷歌健康(Google Health)和加州大学圣克鲁兹分校合作,Benedict Paten及团队开发出了双倍型分型流程(图5)。主要步骤包括通过开源软件minimap2将读长序列比对至所选参考基因组,使用基于SNP的定相过程(候选变异– 基因型分型– 变异定相),最后进行单倍型变异识别获得定相后的候选变异(双倍型)文件。
在单核苷酸变异(SNV)识别中,纳米孔测序在比对定位率低的区域,染色体片段重复区域和碱基序列长度超过250kb方面的表现优于短读长数据
使用人HG002基因组,纳米孔数据在20-22号染色体中的单核苷酸变异(SNV)识别表现要优于使用短读长数据进行的基因分型(图6)。在比对定位率低的区域,由于短读长序列无法很好地定位,长读长有明显优势。以一个染色体片段重复区域为例,短读长数据没有识别出任何SNV,而使用纳米孔读长序列,该区域的所有SNV都被识别出并成功定相。
在均聚物方面的表现与短读长相当
在均聚物(homopolymer stretches)方面,11个碱基对以内的长度里,短读长数据和纳米孔数据的双倍型分型结果相当。在该流程生成的定相block连续性方面,在整个HG002基因组中。其NG50为约1.2Mb,“与此前的数据集相比也非常优秀”。
展望
未来,研究团队希望将Shasta和双倍型分型流程结合起来,以获得完整的、有定相信息的(phase-awared)染色体臂,并通过人类泛基因组计划(Human Pangenome Project)测序来自多样个体的350个人类全基因组,并达到完全定相的、端粒到端粒的质量。(生物谷Bioon.com)
【直播预告】纳米孔测序在人类遗传学和罕见病研究中的应用
【日期】2020/11/19 15:00
http://count.medsci.cn/link/redirect/199d0462698595ba
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
