
Cell:通过非人灵长类疾病模型解析DMD发病早期骨骼肌病理变化的关键机制
来源:生物探索 2024-09-25 10:25
研究结果为DMD发病机制,特别是疾病早期的分子和细胞变化提供了新的见解。揭示了免疫、纤维化以及肌肉干细胞在DMD早期的动态变化,为早期干预和靶向治疗提供了科学依据。
杜氏肌营养不良症(Duchenne muscular dystrophy,DMD)是由法国神经病理学家Guillaume Duchenne首次报道的一种进行性肌肉萎缩疾病,发病率约为1/3500。遗传学研究发现,该疾病是由DMD基因突变导致抗肌萎缩蛋白(dystrophin)缺失的一种X连锁隐性遗传病。患者主要为男孩,通常在2-4岁开始出现走路摔跤等进行性肌肉无力症状,逐渐丧失行动能力,大多在20-30岁因心肺功能衰竭而死亡。由于DMD起病隐匿、病程漫长且缺乏有效的治疗药物,给患者家庭和社会带来了沉重的负担。
针对DMD的基因治疗研究在过去数年中取得了显著进展,已有外显子跳跃和微小dystrophin蛋白补充等药物上市,为患者带来了新的希望。然而,目前已有的小鼠、狗、猪等动物模型无法真实地模拟DMD患者的病程进展,造成对于疾病进展,特别是疾病早期机制的理解仍十分有限。因此在临床应用中依然面临着副作用大、疗效有限、适用范围狭窄以及治疗成本高昂等挑战。为了找到更有效的治疗方法,亟需深入研究DMD在各个疾病阶段的分子和细胞机制。
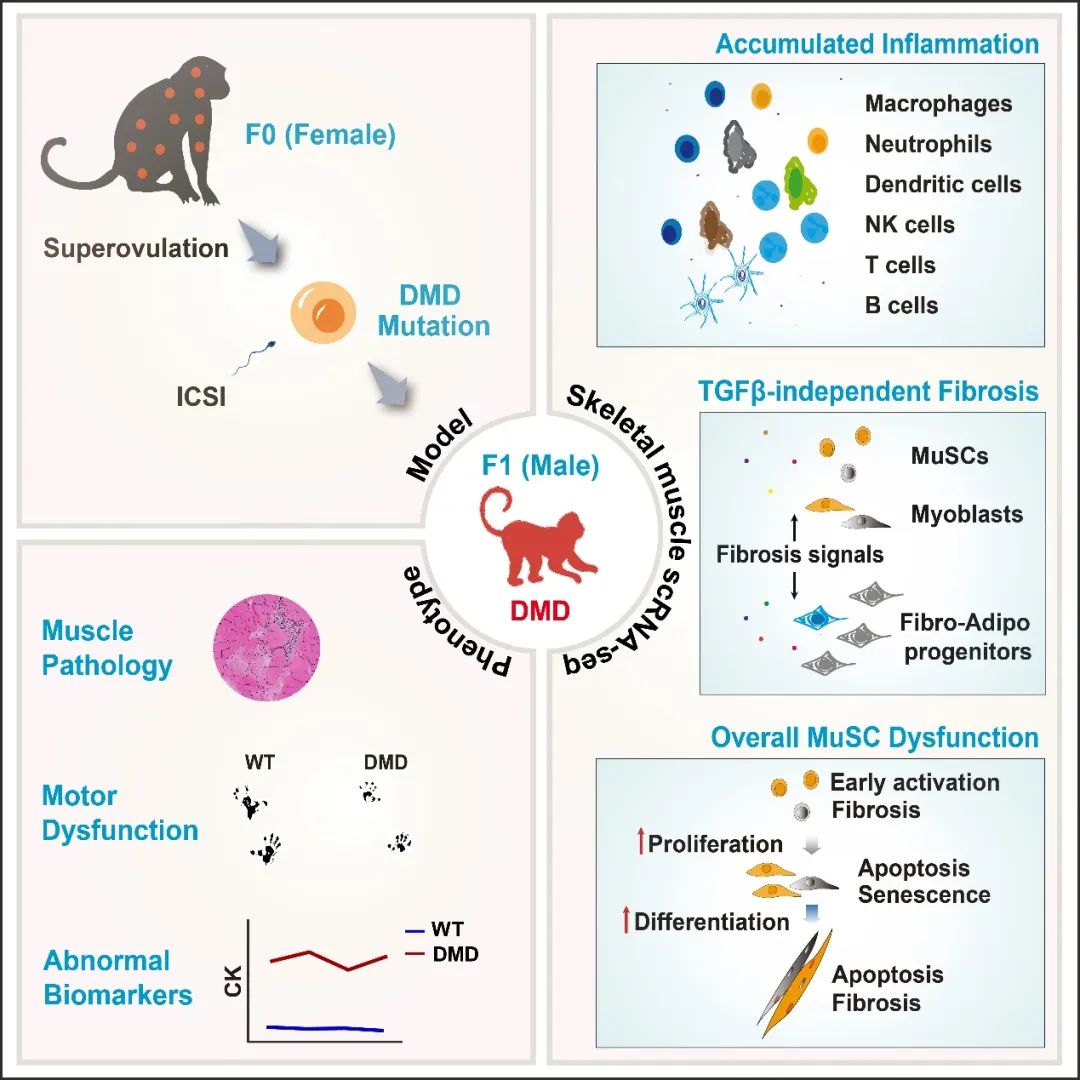
非人灵长类在基因组序列、肌肉结构和功能、生理病理和生长发育进程等方面与人类高度相似,是研究DMD疾病发生发展进程的理想实验动物。昆明理工大学陈永昌/季维智团队自2014年以来在非人灵长类基因编辑和疾病模型构建方面不断取得系列突出进展,2015年在猴中首次实现了对DMD基因的编辑。然而,受限于当时的技术,获得的F0代多为嵌合体状态,难以呈现典型的疾病表型。经过近8年的努力,团队成功培育了F1代DMD猴模型,其中雄性半合子突变个体符合DMD基因型,并展现出与临床患者类似的进展性病理变化。
基于DMD猴模型,陈永昌/季维智团队与胡苹团队将研究重点首先放在了疾病早期的致病机理。由于这一阶段难以对DMD患者进行肌肉活检,且常用的DMD小鼠模型在发病进程和病理表型等方面与临床患者差异巨大,导致对于DMD发病早期的机制仍不十分了解。因此,深入解析DMD发病早期的分子和细胞内在变化,对于探索疾病进程的调控机制和开发早期治疗方案具有重要意义。
陈永昌/胡苹/季维智团队在Cell发表题为Profound cellular defects attribute to muscular pathogenesis in rhesus monkey model of Duchenne muscular dystrophy的研究论文。研究首次发现在DMD的早期阶段,肌肉退化往往首先表现在肌肉组织的微环境和细胞组成上,而这些变化主要涉及单核细胞群,如免疫细胞(Immune cells)、成纤维/成脂肪祖细胞(Fibro-adipogenic progenitors, FAPs)和肌肉干细胞(Muscle stem cells, MuSCs)等。这些单核细胞的动态变化对疾病的进展至关重要。利用单细胞测序技术,研究团队揭示了严重的细胞缺陷是早期肌肉组织病变和再生异常的关键原因,并深入解析了该阶段肌肉组织中单核细胞的变化。

模式图(Credit: Cell)
研究结果为DMD发病机制,特别是疾病早期的分子和细胞变化提供了新的见解。揭示了免疫、纤维化以及肌肉干细胞在DMD早期的动态变化,为早期干预和靶向治疗提供了科学依据。免疫细胞的急剧增加使肌细胞的生存环境恶化,提示抑制炎症反应在DMD治疗中至关重要。此外,DMD猴模型显示FAPs纤维化不依赖于TGFβ通路,为新药研发提供了新的方向。更重要的是,肌肉干细胞功能缺陷导致了肌肉修复障碍,提示DMD是一种干细胞疾病,开发细胞治疗或针对肌肉干细胞进行干预治疗也许是未来研究的重要方向。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
