
利用纳米孔测序研究人体组织样本等位基因效应对转录组结构的影响
来源:NanoporeTechnologies 2020-11-19 12:34
不同的组织和个体之间的基因表达和调控的差异很大,大部分的常见复杂性状相关的基因组通过假定表达和剪接机制来发挥作用,如10%的致病突变都具有已知的剪接效应,RNA测序则能够很好的分析等位基因特异性并解读遗传变异。迄今为止的大多数RNA测序研究都使用短读长RNA测序技术;由于短读长容易出现多重定位的情况,因此很难确定异构体的来源,同时会给定量带来干扰
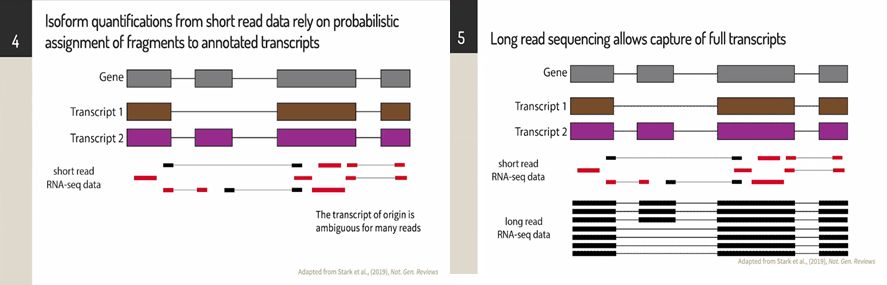
不同的组织和个体之间的基因表达和调控的差异很大,大部分的常见复杂性状相关的基因组通过假定表达和剪接机制来发挥作用,如10%的致病突变都具有已知的剪接效应,RNA测序则能够很好的分析等位基因特异性并解读遗传变异。
迄今为止的大多数RNA测序研究都使用短读长RNA测序技术;由于短读长容易出现多重定位的情况,因此很难确定异构体的来源,同时会给定量带来干扰。相比之下,长读长测序可以捕获到全长转录本和异构体,这可以使定量过程像读长序列计数一样简单。
研究方法与结论
鉴于RNA测序技术需要大量的高质量起始RNA及测序通量等客观原因,长读长RNA测序技术尚未能大规模的应用于人体组织样本,而基于GTEx联盟的生物样本库的丰富数据和纳米孔测序的高通量(单次纳米孔测序运行可获得高达2000万条cDNA长读长序列),来自纽约基因组中心的Dafni Glinos博士及团队对来自43位捐献者的70个人类组织样本(涵盖15种不同的组织)进行纳米孔长读长PCR-cDNA测序,生成了目前规模最大的人类长读长测序cDNA数据集。
利用现有的已定相全基因组数据,研究团队开发出一个将读长序列定位到基因组的工作流程(该流程将在LoRALS工具包中一同发布)。将该流程在全基因组范围应用于所有样本,揭示出跨越4217个基因的11000多个ASE(特异性表达)事件,以及跨越187个基因的350个ASTS(特异性转录本结构事件)事件。
LoRALS全称:长读长等位基因特异性分析,建立于Tuuli Lappalainen实验室的研究之上,发表于:
《Nature Commnication》(DOI:https://doi.org/10.1038/ncomms12817)
《Genome Biology》(DOI:https://doi.org/10.1186/s13059-015-0762-6)
通过比较ASE和ASTS事件发现,并非所有ASTS事件都会以等位基因特异性表达的形式呈现出来,这表明他们能够用这种方法发现细微的转录本使用差异,同时发现四分之一的表达差异可以用转录本结构差异来解释;影响转录本结构的遗传变异通常也会影响表达水平。
使用GTEx联盟定位的大型QTL数据集进行的分析显示,杂合个体的ASE p值比纯合个体低得多,而通过正交验证观察到的ASTS事件反映出了表达和剪接的遗传效应(该项研究由Alexis Battle领导完成,成果预印版本已经发布在《bioRxiv》:DOI: 10.1101/786053)。对这些识别到的显着等位基因特异性转录本结构基因进行检测,发现显着ASTS基因中富集的剪切异常值(outlier),以及罕见的编码杂合变异。通过对GTEx成纤维细胞系进行PTBP1基因敲减PTBP1(一种介导外显子跳跃事件的RNA结合蛋白)来研究ASTS事件,观察到敲减组和对照组之间基因的差异会表现出显着的ASTS事件。
最后,研究团队使用FLAIR分析流程识别到了已知基因的约10万个假定新转录本和大约200个新基因。接下来,在由Michael Snyder实验室的Lihua Jiang牵头的一个项目中,他们计划通过GTEx联盟中同一群个体的质谱数据来验证其中一些新的转录本。初步数据表明,数以千计的转录本在8种主要组织中有表达和使用差异,研究成果发布在bioRxiv(DOI:10.1101/797373)。(生物谷Bioon.com)
【直播预告】纳米孔测序在人类遗传学和罕见病研究中的应用
【日期】2020/11/19 15:00
http://count.medsci.cn/link/redirect/199d0462698595ba
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
