
药品监管制度变化对中国创新药可及性的影响
来源:医药魔方 2023-04-25 12:05
自2015年以来,国家药监局推出了一系列的药品监管改革措施。为了衡量这些改革的影响,复旦大学药学院教授、上海市药物研发协同创新中心主任邵黎明团队研究了2015–2020年间FDA批准的新药
自2015年以来,国家药监局推出了一系列的药品监管改革措施。为了衡量这些改革的影响,复旦大学药学院教授、上海市药物研发协同创新中心主任邵黎明团队研究了2015–2020年间FDA批准的新药在中国的可及性(数据来源FDA、NMPA以及药企官网)。这也是对他们之前使用2004–2014年数据进行类似分析(doi:10.1038/nrd.2016.200)的更新。相关文章近期发表在Nature Reviews Drug Discovery上。

2015年至2020年,FDA共批准了200个新分子实体(NME)。同期,62种新药在中国获批,其中包括28种NME、17种新生物制品和17种中药。
在美国批准的200项NME中,截至分析截止日期2022年2月1日,55项(28%)也在中国获批(时间框架与邵教授团队之前的分析方法一致)。图1显示了美国和中国批准的NME的数量。补充表1和补充表2中提供了在美国和中国批准的所有药物的清单,包括它们的治疗领域以及它们在两国的提交和批准日期。
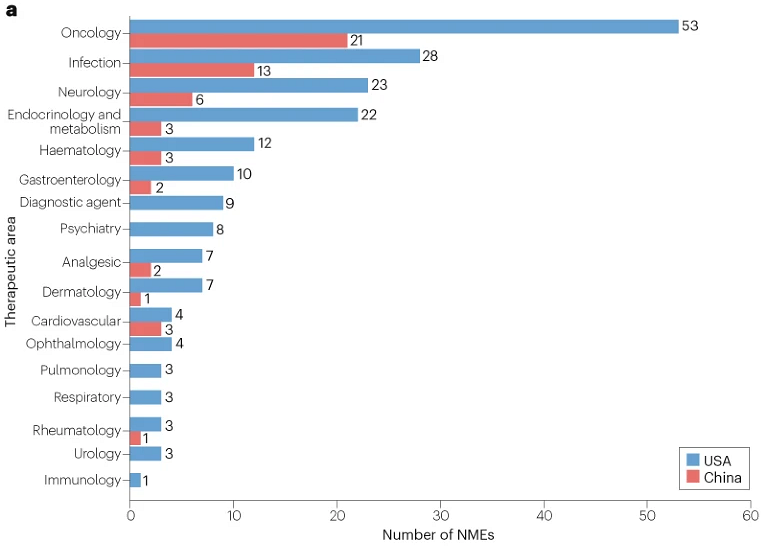
图1 | 2015–2020年间在美国也在中国批准的新药分析(按治疗领域划分)
在此期间,针对癌症和传染病的药物是在中美两国都批准的NME中最常见的。值得注意的是,2015–2020年间,FDA批准的53种针对肿瘤适应症的NME中有21种(40%)在中国获得批准,而相应在2004–2014年间,58种NME中有14种(24%)获得批准。这一比例的提高可能是由于NMPA引入了适应症团队审查系统,该系统成立于2016年,始于肿瘤学适应症。中国癌症患者的大量未满足需求也加快了中国进口肿瘤药物的审批。
然而,在其他治疗领域很少有FDA批准的NME在中国获批,而且这个“小比例”与之前在2004–2014年的观察结果基本一致。不过,2015–2020年,美国FDA在内分泌和代谢治疗领域批准的22种NME中,只有3种(14%)在中国获得批准,而2004–2014年为32%(12/38)。
此外,2015–2020年间美国批准的200项NME中,93项(47%)被批准用于治疗罕见病,其中23项也在中国获批(图2),总体比例相似(23/55;42%)。
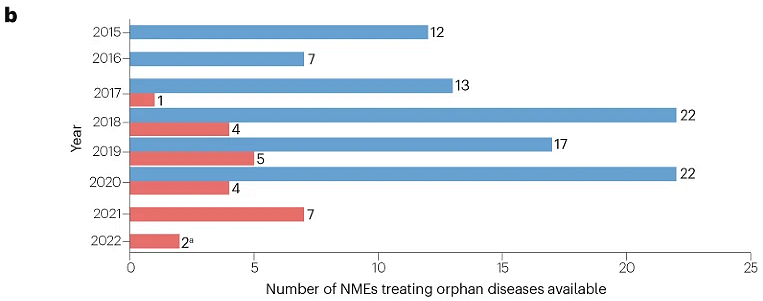
图2 | 200种NME中,各年在美国和中国批准用于罕见病(获FDA孤儿药资格认定 )的数量,中国批准的截止日期为2022年2月1日(蓝色为美国,红色为中国)
对于2015–2020年间获得FDA以及NMPA批准的NME,中美两国批准的平均滞后时间为2.2年,短于2004–2014年间观察到的3年滞后时间(图3)。滞后时长缩短是由近年来NMPA的批准推动的,2018–2020年的平均滞后时间进一步缩短为1.2年。只有一种创新药物首先在中国获得批准:治疗慢性特发性便秘的普芦卡必利(中国早6年批准)。两种创新药物几乎同期在中美获批:用于治疗慢性心力衰竭的伊伐布雷定和用于诱导和维持成人手术镇静的瑞马唑仑(中国晚15天批准)。然而,一些新药在中国的批准时间比美国晚很多。例如,抗真菌药物艾沙康唑在美国首次批准约7年后在中国获得批准,抗癌药物索立德吉在美国批准6年后在中国获批。
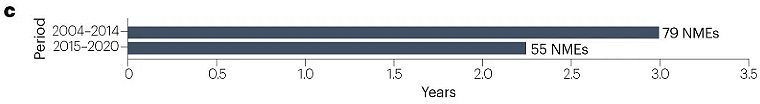
图3 | 美国和中国NME批准之间的时间滞后
邵教授团队还计算并比较了从新药申请(NDA)到每种药物在美国和中国都获得批准的持续时间。在中国,大多数新药从NDA提交到批准平均需要大约1年或更短的时间,这与美国NDA审查时间接近。
国家药监局自2015年以来的监管改革大大提高了药品审评和审批过程的透明度和效率。以下附上2015年以来助力加快新药审批的几项重大监管改革政策:
-
为了加快临床急需的国外新药的审评和批准,2015年3月,NMPA规定进口药物可以在中国进行多中心临床试验。2017年6月,NMPA加入了人用药品技术要求国际协调理事会(ICH),并引入了药物研发和注册的全球标准(如ICH E5和ICH E17)。
-
2017年10月,NMPA发布了《关于调整进口药品注册管理有关事项的决定》,规定在中国进行国际多中心药物临床试验,允许同步开展I期临床试验。一个例子是 Epclusa(索磷布韦+维帕他韦),用于治疗成人和3岁及以上儿童的慢性丙型肝炎。Epclusa于2018年5月(FDA批准23个月后)在中国迅速获得批准,主要基于五项多中心III期试验。
-
2018年7月,NMPA发布了《国家药品监督管理局关于调整药物临床试验审评审批程序的公告》,对临床试验申请启动了60天审批制度,也被称为临床试验默示许可制度。2018年11月正式实施后,IND的平均审批时间从6个月或更长时间稳步下降到60个工作日内。
-
同样在2018年7月,NMPA发布了《接受药品境外临床试验数据的技术指导原则》,以加快临床急需的海外新药的审批。2018年10月,NMPA与国家卫生健康委员会共同发布了《临床急需境外新药审评审批工作程序》。2020年,CDE在3个月的规定时限内完成了所有临床急需的13种罕见病治疗药物的技术审评,其他26种急需药物的审评在6个月内完成,减少了进口新药审批的时间滞后。
-
最后,2020年7月,NMPA开始实施新修订的《药品注册管理办法》,进一步优化了审评审批流程,鼓励制药公司基于临床实用性开发新药。2020年10月,CDE发布了《境外已上市境内未上市药品临床技术要求》,列出了3类可以减少或豁免临床试验的境外原研药。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
