
《自然·遗传学》:超百万人跨种族全基因组研究,发现全新肺癌易感基因位点!
来源:奇点糕 2022-10-05 10:18
肺癌的发生是一个相对复杂的过程,包括吸烟、遗传因素和体细胞突变积累等在内的多种因素均能够驱动肺癌的发生和发展[1]。
肺癌的发生是一个相对复杂的过程,包括吸烟、遗传因素和体细胞突变积累等在内的多种因素均能够驱动肺癌的发生和发展[1]。
在此前的研究中,通过全基因组关联研究(GWASs),研究人员们已经鉴定出了许多肺癌易感位点,这对肺癌的筛查和诊断产生了很大的帮助。然而,由于这些研究中所采用的样本大多是有着欧洲血统(EUR)的人群,样本内部存在很大的遗传同质性,因而可能有许多罕见的、与人种相关的遗传突变位点尚未被揭示[2]。
近日,一个由美国贝勒医学院领导的国际团队在《自然·遗传学》上发表了一项重要研究成果,通过对来自欧洲、东亚和非洲的样本(61047例病例和947237例对照)进行跨种族的全基因组荟萃分析(GWMA),研究人员发现了多个此前未被鉴定的新肺癌易感位点[3]。

论文首页截图
在过去20多年的研究中,通过GWASs等分析手段,研究人员们已经确定了40余个与肺癌风险直接相关的易感性位点。基于这些易感位点进行估测,人群中遗传因素对于肺癌发病的影响大致在8%到21%之间,也就是说,肺癌在引发原因(包括吸烟、遗传因素和体细胞突变积累等)上存在着较大的异质性。
为了进一步探究不同人种间遗传性因素对肺癌引发的影响,研究人员们整合了多个此前已经完成的GWASs分析数据集,并进行了跨种族的GWMA分析,全面的描述了常见和罕见的肺癌遗传易感性位点。
在这项分析中,研究人员们共计纳入了来自12个不同祖先群体的70156个病例样本,在这之中,有74%的样本(51961例)被推断为具有欧洲血统(EUR),18%(12434例)具有东亚血统(EAS),8%(5766例)具有非洲血统( AFR)。
为了避免不同祖先群体间数据处理,特别是在小样本量和分析罕见变异时产生的偏差,研究人员们首先按照EUR、EAS和AFR对样本进行了分层。然后再结合由单倍型参照联合体(HRC)中抽取的32470份对照样本进行跨种族GWMA荟萃分析[3]。这一研究阶段在后续描述中被称为发现阶段(discovery phase)。
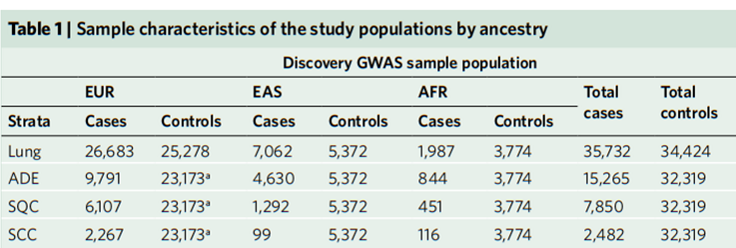
图1 发现阶段(discovery phase)所分析样本的来源
最终,研究人员共计鉴定出40个肺癌相关性单核苷酸多态性(SNPs)位点(按照显著性水平P_BE1<1.25×10-8的标准,对应到下图为红色基线以上位点)。
按照病理组织学分类,其中的15个与整体性肺癌(Overall lung cancer)发病相关,14个与肺腺癌(ADE)相关、9个与肺鳞状细胞瘤(SQC)相关,2个与小细胞肺癌(SCC)相关。
另外,在这些位点中,共有9个SNP突变(下图中标注为蓝色字体)是此前研究中所未被鉴定的。
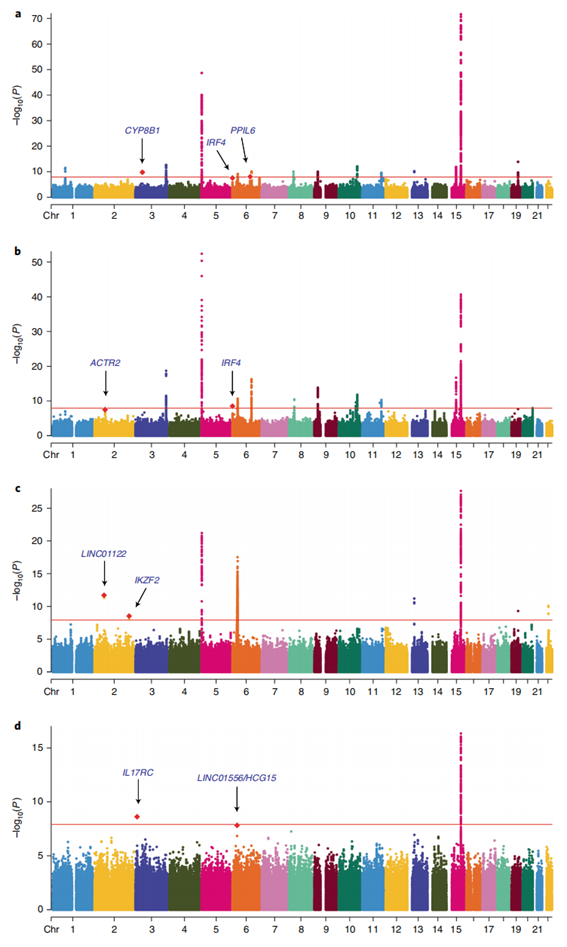
图2 对不同病理组织学亚型肺癌进行跨种族GWMA荟萃分析得到的曼哈顿图
为了确认上述跨种族GWMA荟萃分析结果,区别于上述发现阶段(discovery phase),研究人员又进一步采用了一个由938128个个体组成的大队列(数据来源于7项已发表研究,样本既包括肺癌病例,也包括对照样本)进行验证。该队列的数据来源是种族特异性的(即分别来源于EUR、EAS、AFR血统),且未被纳入上述在发现阶段(即图1所示样本)所中进行的跨种族GWMA荟萃分析。
在这里,研究人员总计对发现阶段所鉴定出的45个推测与肺癌发病存在相关性的SNP突变进行了验证。
结果发现,其中9个与整体性肺癌相关、10个与ADE相关、6个与SQC相关、1个与SCC相关的SNP位点,是在此前研究中已被证实的;另外,有4个与整体性肺癌相关、5个与ADE相关以及1个与SQC相关的新鉴定SNP位点,位于此前已被鉴定的突变位点周围(±500kb)。
总之,综合验证结果表明,跨种族的GWMA荟萃分析不仅可以稳定地检测到此前已被报道的肺癌相关SNP位点,还能有效地检测到一些新的突变位点。
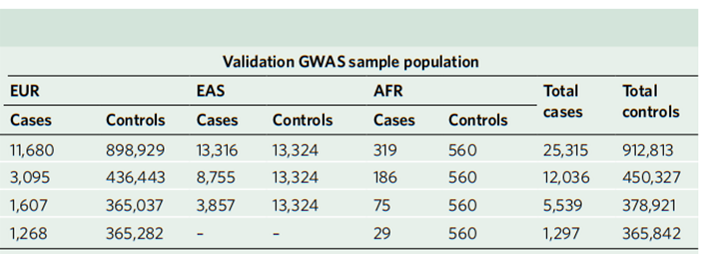
图3 验证阶段(validating phase)所分析的样本来源
除了位于此前已被报道过的易感位点(susceptibility loci)周围的新信号(指新鉴定的SNP突变)或已知信号,这项跨种族GWMA荟萃分析还发现了众多新的肺癌相关易感性位点。
在这些位点中,突变rs12203592(位于IRF4基因的一个内含子上)在此前的研究中已被报道与众多色素沉着特征、血细胞特征、皮肤鳞状细胞癌和戒烟行为相关,这表明该突变位点存在着多效性。
另外,基因PPIL6与血液性状的关联已在EUR人群的种族特异性研究以及跨种族分析中被证实,而此次发现,PPIL6基因上的一个内含子突变rs17534632同时与EUR人群、EAS人群的肺癌风险增加有关。
总之,这些特征可能表明了新鉴定易感基因位点的潜在致病机理。
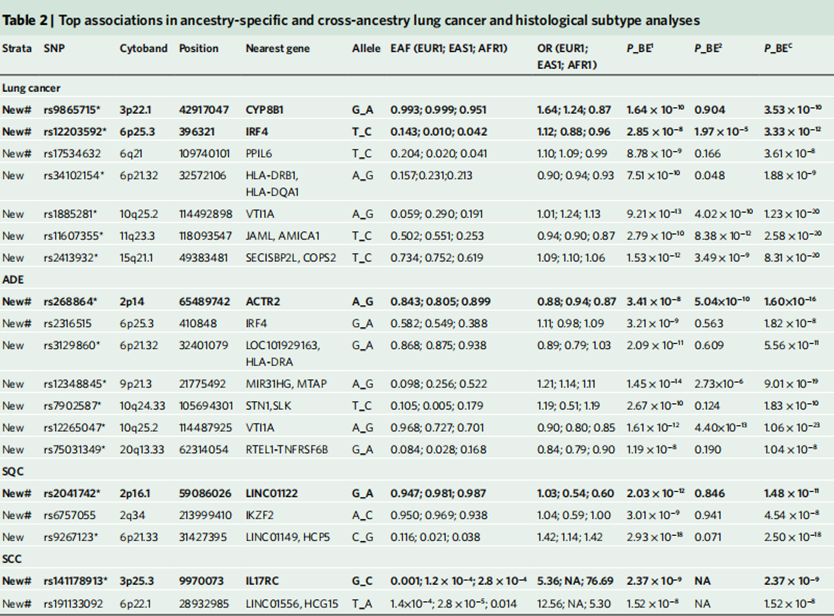
图4 新鉴定的肺癌易感基因位点
在进一步分析中,作者使用了基于表达数量性状位点(eQTL)的分析对候选易感基因位点进行功能表征。
结果表明,有63%的候选基因能够改变肺成纤维细胞的内源性DNA损伤水平,其中,本项研究中新鉴定出的易感基因位点(IRF4、ACTR2、PPIL6和AK9)均能够促进或抑制内源性DNA损伤。
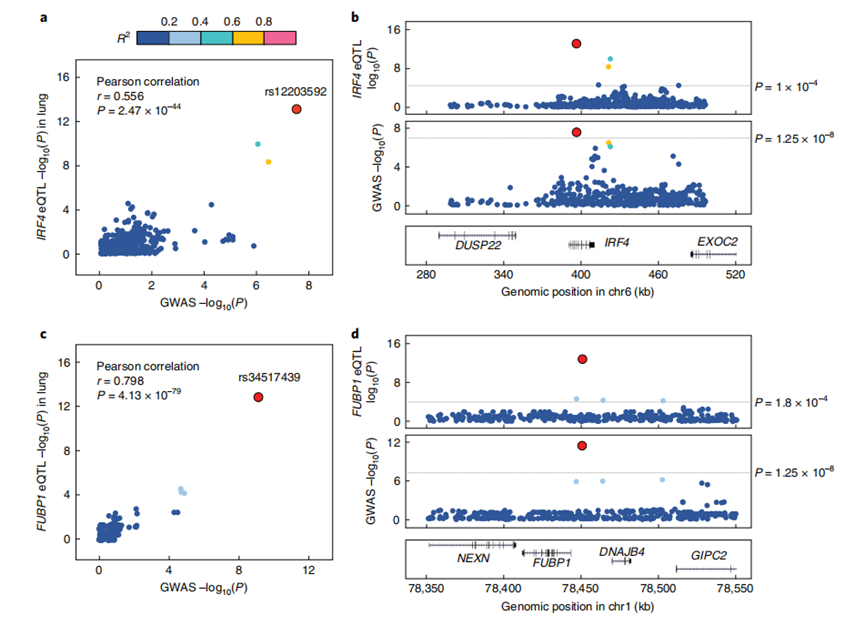
图5 跨种族GWAS分析所鉴定的优先易感基因的功能验证
接下来,为了进一步表征候选肺癌易感基因的优先级,研究人员们又进行了基于细胞的DNA损伤水平分析。
目前,已经有越来越多的基因被证实可通过各种直接和间接的机制促进DNA损伤,而DNA损伤的积累则会进一步导致基因的突变和癌症的引发。在这里,研究人员假设,GWAS鉴定的肺癌相关风险基因也能够通过增加内源性DNA损伤和基因组不稳定性来促进癌症的进程。
具体而言,研究人员在永生化的人肺成纤维细胞中进行了相关候选基因的敲除和/或过表达,通过这一过程来模拟肺癌产生过程中候选基因的表达变化,然后,再通过γ-H2AX的水平来评估DNA损伤的水平。
最终,在所挑选的19个基因中,7个过表达基因(IRF4、AK9、CYP21A、DCBLD1、SECISBP2L、CCDC97和FUBP1)和2个敲除基因(PPIL6和ACTR2)表现出显著的DNA损伤增加。
在这里以IRF4为例,当其过表达时,细胞表现出增加的DNA损伤水平,而肺组织中IRF4的内源性表达水平也确实高于其它组织,这可能反映了其促进肺癌风险的潜在机制。另外,ACTR2基因在敲除时表现为DNA损伤增加,而在过表达时则表现为DNA损伤的减少,这表明其可能存在潜在的保护作用。
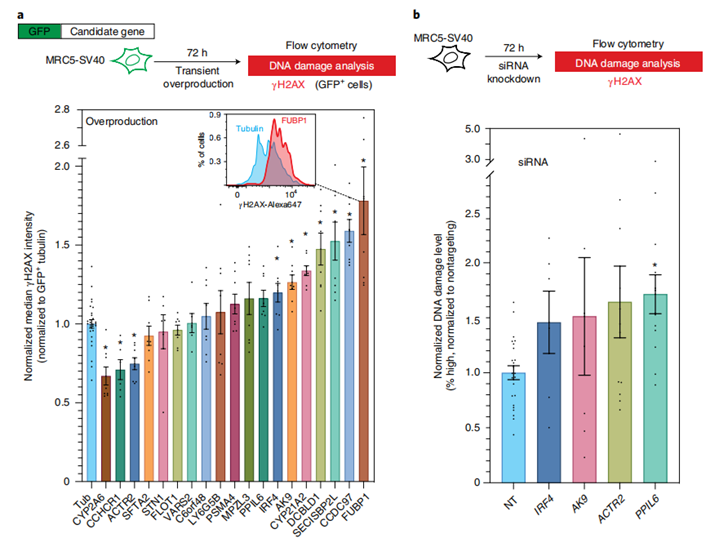
图6 肺癌易感基因对细胞内DNA损伤水平的影响
总的来说,这项研究通过对大规模种族特异性样本进行跨种族GWMA荟萃分析,鉴定和完善了肺癌相关的易感基因位点,这将进一步帮助阐明肺癌的病因和发病机制。
未来对相关易感性位点的识别和筛查,也将大大促进肺癌的早期诊断、靶向治疗和预后改善。
参考文献:
[1]Sampson JN, Wheeler WA, Yeager M, et al. Analysis of Heritability and Shared Heritability Based on Genome-Wide Association Studies for Thirteen Cancer Types [published correction appears in J Natl Cancer Inst. 2016 Apr;108(4). pii: djw106. doi: 10.1093/jnci/djw106]. J Natl Cancer Inst. 2015;107(12):djv279. Published 2015 Oct 12.
[2]Asimit JL, Hatzikotoulas K, McCarthy M, Morris AP, Zeggini E. Trans-ethnic study design approaches for fine-mapping. Eur J Hum Genet. 2016;24(9):1330-1336.
[3]Byun J, Han Y, Li Y, et al. Cross-ancestry genome-wide meta-analysis of 61,047 cases and 947,237 controls identifies new susceptibility loci contributing to lung cancer. Nat Genet. 2022;54(8):1167-1177.
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
