
Nat Commun | 浙江大学王继荣/吴希美等合作发现SHH信号在髓母细胞瘤中起重要作用
来源:生物探索 2024-02-09 10:52
该研究证明了SHH通过使p38α失活来使hGLI1在Ser937上去磷酸化(mGli1在Ser941上),从而稳定GLI1来诱导SHH信号的转录输出。
浙江大学王继荣、吴希美及浙大城市学院曾玲晖共同通讯在Nature Communications (IF 16.6)发表题为“Phosphorylation of human glioma-associated oncogene 1 on Ser937 regulates Sonic Hedgehog signaling in medulloblastoma”的研究论文,该研究发现SHH以Smoothened依赖性方式使p38α(MAPK14)失活,相反,p38α直接在Ser937/Ser941上磷酸化GLI1(人/小鼠),从而诱导GLI1的蛋白酶体降解,并否定SHH信号的转录。
Gli1S941E系列功能丧失敲入显著降低平滑M2转基因诱导的自发性MBSHH的发生率和严重程度,而Gli1S941A功能获得性敲入表型Gli1转基因在皮肤中引起BCC样增殖。相应地,磷酸化Ser937-GLI1(GLI1的一种不稳定形式)与MBSHH患儿的总生存率呈正相关。总之,这些发现表明SHH诱导的p38α失活和随后的GLI1 去磷酸化和稳定在控制SHH信号传导方面的作用,并可能为未来干预MBSHH和BCC提供途径。

Hedgehog (HH)信号通路对胚胎发育和肿瘤发生至关重要。哺乳动物HH配体由Sonic Hedgehog(SHH)、Indian Hedgehog(IHH)和Desert Hedgehog(DHH)组成。通过将HH配体与patched受体(PTCH)结合,可以解除对HH通路的辅助受体smoothened (SMO)的抑制,进而允许胶质瘤相关癌基因(GLI)家族转录因子GLI1、GLI2和GLI3诱导靶基因的转录。GLI2 和 GLI3 转录因子以蛋白酶体依赖性方式加工以产生转录激活因子或阻遏因子,而GLI1不仅是转录因子,也是HH信号通路的靶蛋白,通常被认为是转录激活因子,不需要蛋白酶体依赖的加工。融合抑制因子(SUFU)是一种细胞质蛋白,与所有三种GLI蛋白相互作用,并作为GLI转录因子的抑制因子。
髓母细胞瘤(MB)是儿童最常见的小脑恶性肿瘤之一,约占所有儿童脑肿瘤的20%。MB有四个分子亚型,包括WNT组、SHH组、3组和4组,其中MBSHH是最常见的形式。基底细胞癌(BCC)是最常见的皮肤癌形式,也是所有癌症中最常见的形式。HH相关的基因改变常发生于MBSHH(87%)和BCC (85%), Ptch1和Sufu的功能缺失突变以及Smo和Gli2的功能获得性突变通常见于MBSHH和BCC,所有这些突变均导致GLI1水平上调。因此,GLI1被认为是这两种癌症发病率和严重程度的关键效应因素。
在单倍不足的Ptch1+/-小鼠中,消融Gli1显著减少了MBSHH的自发形成。主要由于SHH信号通路的重要性,SMO抑制剂vismodegib和sonidegib已被批准用于治疗BCC,并正在进行MBSHH药物治疗的临床试验。尽管vismodegib对携带Ptch1或Smo突变的MBSHH有效力和疗效,但对于产生代偿机制并将SHH信号恢复至受体后水平的MBSHH无效,而且对Sufu或Gli基因改变的MBSHH几乎无影响。因此,在受体后水平,特别是在GLI1水平,识别出能够阻断信号传导的方法和分子,可能是这些疾病治疗干预的更好选择。
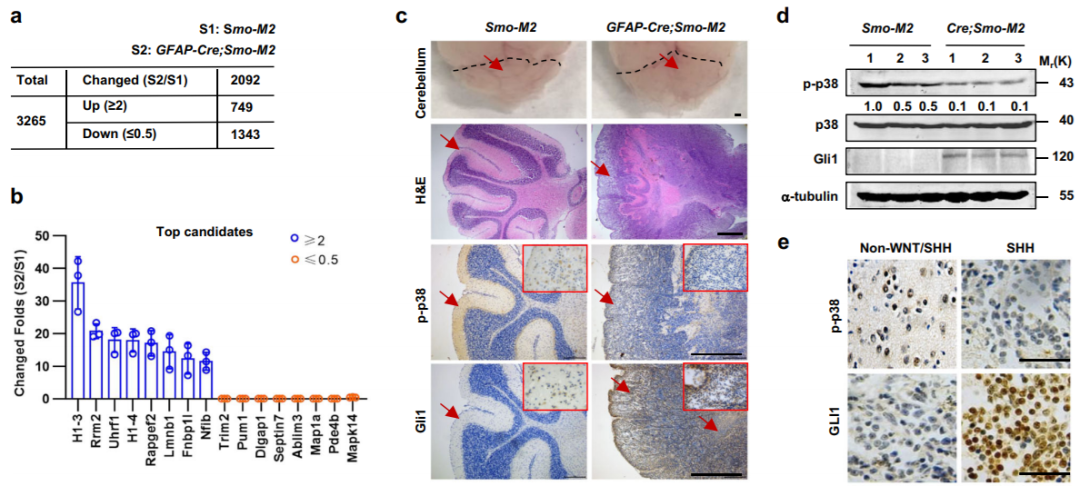
SHH信号的激活使p38失活以诱导Gli1蛋白(Credit: Nature Communications)
该研究证明了SHH通过使p38α失活来使hGLI1在Ser937上去磷酸化(mGli1在Ser941上),从而稳定GLI1来诱导SHH信号的转录输出。因此,Gli1S941E的功能丧失性突变降低了SMO诱导的组成型活性MBSHH的发生率和严重程度,而Gli1S941A的功能获得性突变表型复制了Gli1转基因,导致皮肤BCC样增殖。重要的是,p-Ser937-GLI1水平与MBSHH患者的总生存期呈正相关。p38α介导的GLI1 Ser937/Ser941位点的磷酸化与MBSHH和BCC的发病和严重程度密切相关,因此干预GLI1的这一关键磷酸化位点可能为这些疾病的药物治疗提供新的途径。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
