
Nature子刊:“数据+知识+AI” ,郑明月团队解锁新靶标药物虚拟筛选,为基于结构的药物设计提供新线索
来源:生物世界 2024-06-09 13:10
科研人员提出了一种信息感知注意力机制,用于整合不同信息中的相互作用,这些信息包括包括:1)等变几何信息;2)化学结构信息;3)经验相互作用信息。
中国科学院上海药物研究所郑明月课题组在 Nature 子刊 Nature Machine Intelligence 上发表了题为:Generic protein–ligand interaction scoring by integrating physical prior knowledge and data augmentation modelling 的研究论文。
该研究构建了一种通用蛋白质-配体相互作用评分方法——EquiScore。在药物虚拟筛选场景和先导化合物优化场景中,EquiScore对训练未见的新靶标表现出了良好的泛化性能。此外,EquiScore的可解释性分析为基于结构的药物设计提供了有价值的线索。

该研究主要从两个方面来提高深度学习评分函数对新蛋白的预测能力。首先,科研人员收集了更多的阳性样本,并使用重对接来生成更多样的阳性样本。同时,使用交叉蛋白对接,分子生成模型来生成更多具有欺骗性和多样性的诱饵分子,以减少构建训练数据集时可能出现的类似物偏差(analog bias)、数据分布偏差(data distribution bias)以及人工富集偏差(artificial enrichment bias)(图1)。其次,团队提出了一种异质图构建流程,可以通过引入新的节点和边来整合分子间相互作用的物理先验信息。同时,科研人员提出了一种信息感知注意力机制,用于整合不同信息中的相互作用,这些信息包括包括:1)等变几何信息;2)化学结构信息;3)经验相互作用信息。研究团队通过使用新构建的数据集和等变异质图网络来训练最终的评分模型EquiScore。
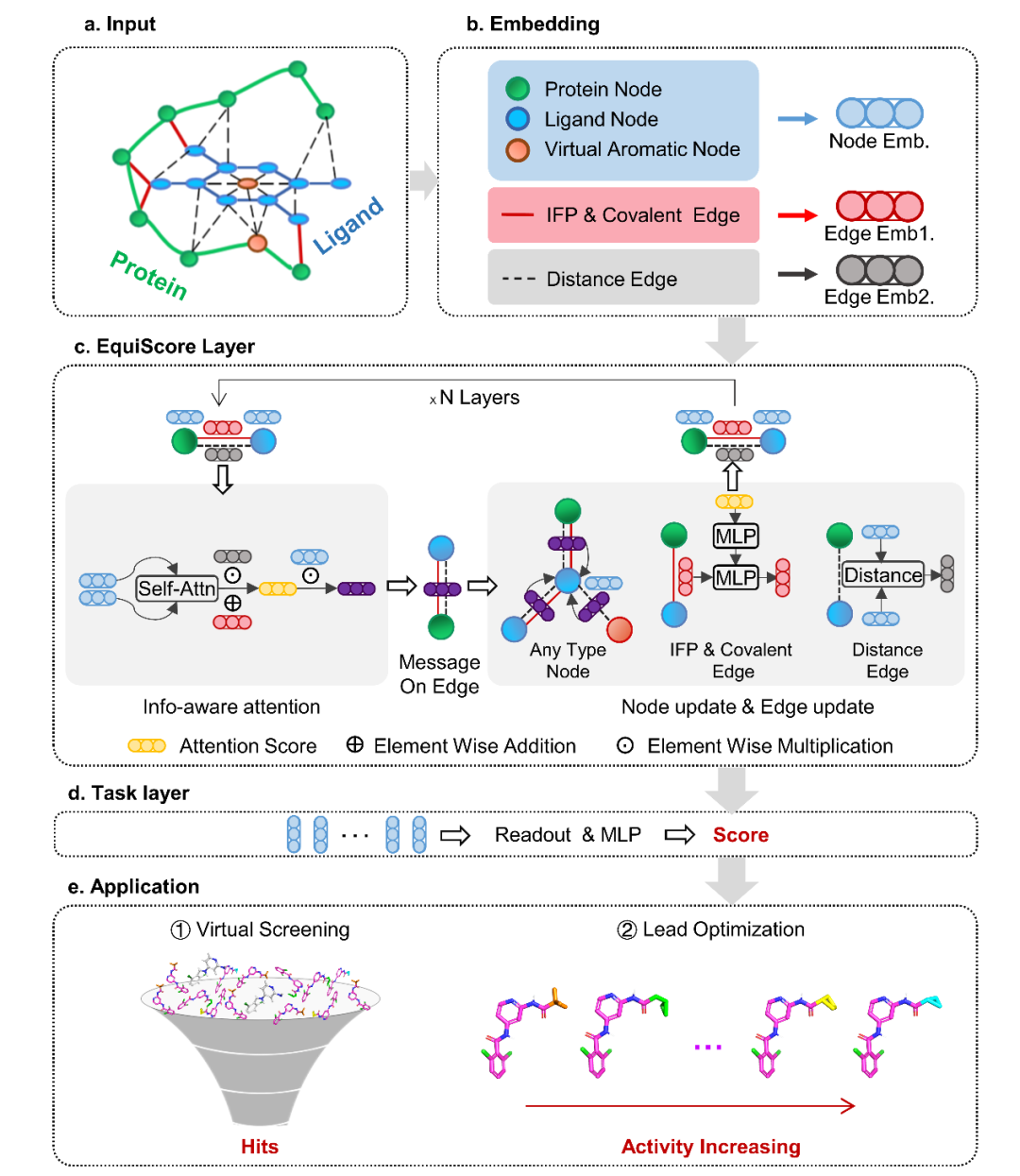
图1. EquiScore 架构图。a:构造异质图引入物理先验信息。b: 嵌入层。c: EquiScore层。d:任务层。e: 应用场景。
在训练集中未见过的蛋白质上的虚拟筛选能力能够更好的反映评分方法在实际应用中的泛化性能。为了进行充分的比较,研究团队选择了21种不同的评分方法作为基准。最近报道的所有方法都是基于PDBbind数据集训练得到的,该数据集与外部测试集具有高度的“软重叠”,即很多蛋白都是在训练过程中模型已经见过的。
为了进一步检查这种数据泄漏是否会导致性能高估,研究团队将外部测试集涉及“软重叠”的数据进行了去重,并对所有方法的结果进行重新评估。在DEKOIS2.0测试集上的分析结果显示(图2),EquiScore的排序能力和富集能力都排在第一或者第二。值得注意的是,当只考虑训练过程中未见过的蛋白时,EquiScore 在所有的结果上都排在第一位。以上结果表明,在严格的测试下,EquiScore的综合排序能力超过了现有的方法。此外,EquiScore对新蛋白的富集能力超过了传统评分方法和深度学习方法。
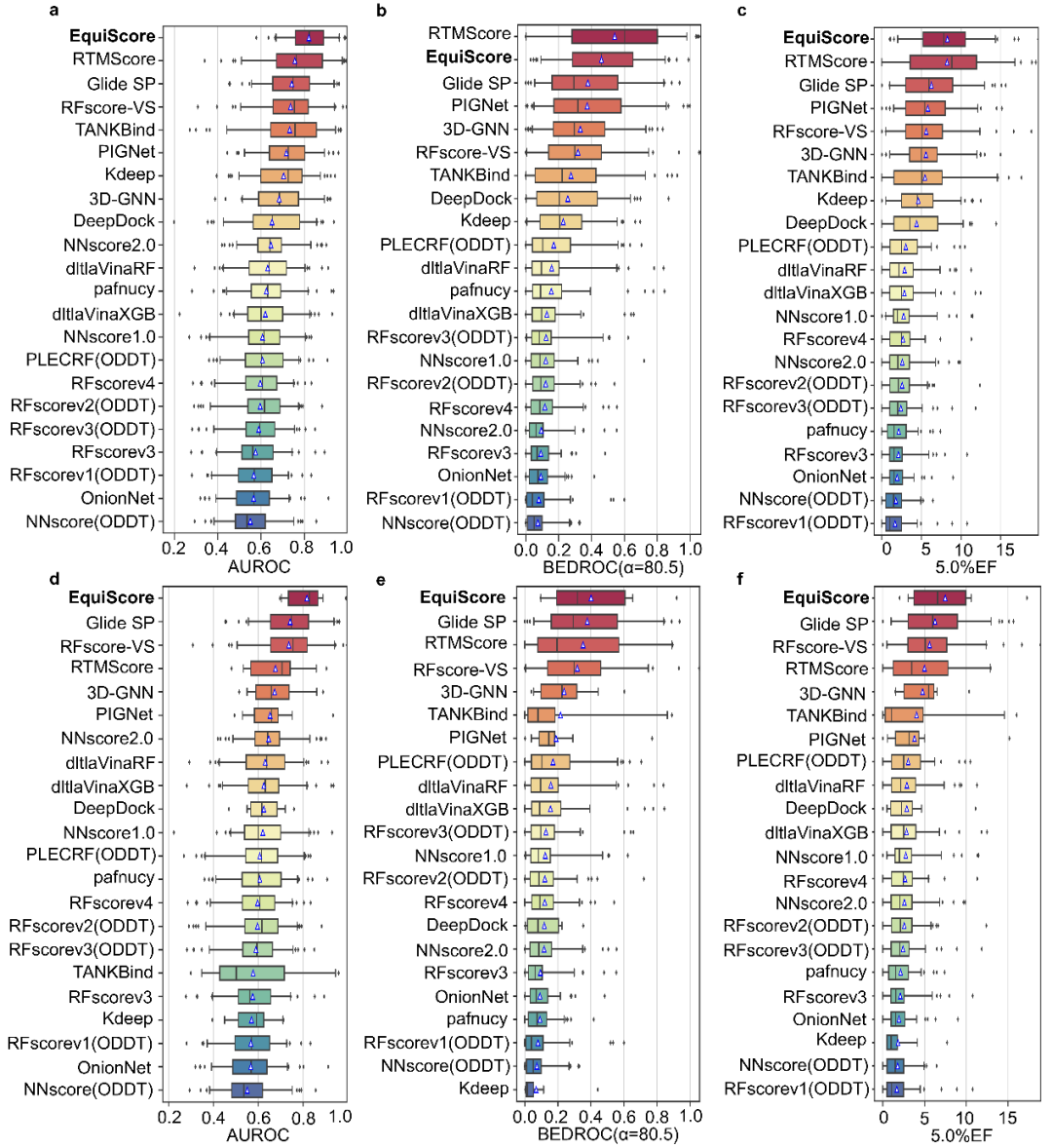
图2. 在DEKOIS2.0上对22种评分方法进行评估。a、d: AUROC, b、e: BEDROC (α = 80.5)和c、f: 5.0% EF。箱形图中的蓝色三角形代表每个箱子的平均值。所有方法按其平均值排序
同时,研究团队还在外部的先导化合物优化数据集上比较了EquiScore与其它方法对结构类似物的活性排序能力; 使用不同的对接方法生成蛋白-配体复合物结合构象,进一步评价EquiScore作为评分方法的鲁棒性。最后,研究团队还分析了模型的可解释性。
浙江大学与上海药物所联合培养博士研究生曹端华,国科大杭州高等研究院硕士研究生陈庚为论文共同第一作者。中国科学院上海药物所郑明月研究员为本文通讯作者。该研究得到了国家自然科学基金、国家重点研发计划、上海药物所与上海中医药大学中医药创新团队联合研究项目、中国科学院青年创新促进会会员项目、上海市科技重大专项资助。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
