
《自然·生物技术》:从2天到几十秒!MIT科学家发明癌症驱动突变挖掘算法
来源:奇点糕 2022-07-19 15:50
研究人员基于概率深度学习方法开发的Dig算法可快速、准确地在测试基因组中寻找潜在的突变基因,其在构建中性突变率模型和识别候选驱动基因方面的强大性能,突出了深度学习在该类研究中的广阔前景。
物竞天择,适者生存。
这是自然界乃至人类社会普遍存在的规律。
从这一角度来说,肿瘤细胞正是得益于一些具有“正向选择”功能的驱动突变,这类基因突变往往可以起到促进细胞生长及侵袭、抵抗死亡等作用,从而赋予肿瘤细胞增殖优势[1]。
识别这些突变的驱动基因一直以来都是了解肿瘤发病机制,及寻找相对应治疗策略的重要一环。
然而,肿瘤基因组中存在众多中性突变(对肿瘤的发生发展无关键作用),且不同组织来源的肿瘤其突变具有一定的特异性[2],如何判断一个突变是驱动突变还是中性突变一直以来都是肿瘤领域研究的热点与难点。
近日,来自麻省理工学院Bonnie Berger教授团队和哈佛医学院Po-Ru Loh教授团队携手,通过深度学习的方法绘制了千碱基级分辨率的癌症特异性体细胞突变率图谱,并在此基础上开发了可快速、精准识别肿瘤基因组中任意位置驱动突变的方法(Dig算法)。
研究人员使用该算法对肿瘤非编码区的驱动突变进行了探索,发现了内含子中隐蔽剪接单核苷酸变异,及5’非翻译区突变在部分驱动基因(如TP53)中起到关键作用,相关研究发表于《自然·生物技术》杂志上[3]。

论文首页截图
通常识别驱动突变的方法主要有两类,一是基于癌症驱动突变比中性突变频率较高的频率算法,二是基于突变基因所编码蛋白有害性评价的基因功能算法。对于第一种算法,识别过量突变的关键在于一个准确的体细胞中性突变率的模型。
为了构建特定类型肿瘤的全基因组体细胞突变率模型,研究人员采用概率深度学习模型,通过识别以下两个特征来进行计算:由表观遗传学特征如染色质可及性等导致的千碱基级变异,以及由DNA序列改变如紫外线暴露等导致的碱基对级变异。
通过获取到的PCAWG(泛癌数据库)、Roadmap Epigenomics(表观遗传学数据库)、ENCODE(转录调控数据库)以及参考基因组的数据,研究人员应用Dig算法构建了37种癌症类型的突变率图谱和推断的核苷酸突变偏向性。
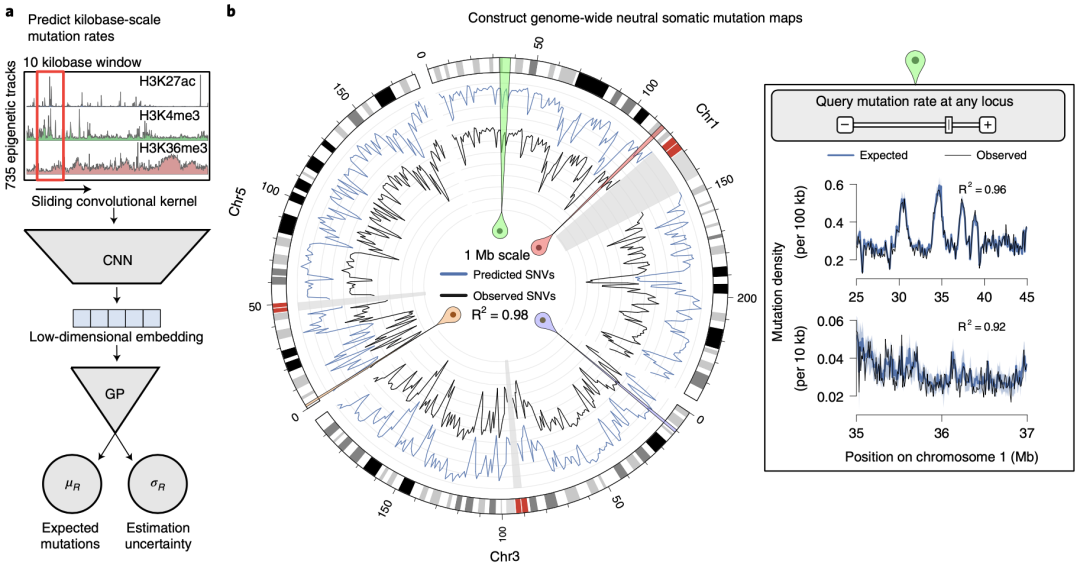
构建特定类型肿瘤的全基因组体细胞突变率模型
通过比对Dig预测的突变率及实际的突变率,Dig成功预测10-kb区域内单核苷酸变异(SNV)的中位数为77.3%,在1-Mb区域内的中位数为94.6%。由于Dig具有识别局部表观遗传结构的能力(如活跃的转录起始点),并将这些结构与突变率联系起来,其识别突变的效率及准确性优于同类的其他模型。
在构建的体细胞突变率模型具有优异准确性的基础上,Dig识别肿瘤驱动基因的效率自然也不遑多让。相比于其他用于识别驱动突变的算法,Dig在全基因组或全外显子测区的样本中显示出相同或更好的效率。
值得一提的是,使用Dig识别潜在的驱动基因比现有方法快1-5个数量级,Dig只需要不超过90秒,就可抵得上现有方法10分钟到超过2天的计算。
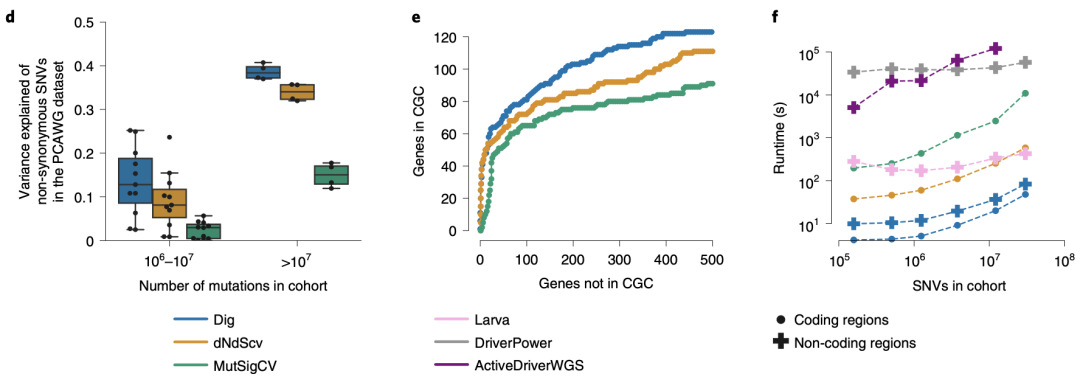
相比于同类型算法,Dig在预测体细胞突变路及识别肿瘤驱动基因的效率更好
对于编码区的驱动突变,已有很多算法进行研究,因此,研究人员着重利用Dig对非编码区的驱动突变进行研究,这也是Dig算法的一大优势,先前的许多算法都仅适用于寻找编码区的驱动突变。
首先,研究人员利用Dig算法来量化肿瘤中可能存在于基因外显子和内含子中的隐蔽剪接SNV(经典剪切位点外的体细胞突变并可致可变剪切)的程度。
通过计算发现,在所选择研究的肿瘤抑制基因集中,隐蔽剪接SNV的发生频率要高于预期(P<0.001),且主要集中在内含子中。在肿瘤抑制基因集中,隐蔽剪接SNV约占到所有潜在驱动SNV的4.5%,略低于经典剪切SNV的频率(7.4%)。在12种肿瘤中,有7个肿瘤抑制基因具有显著的内含子隐蔽剪接SNV负担,如TP53和SMAD4。
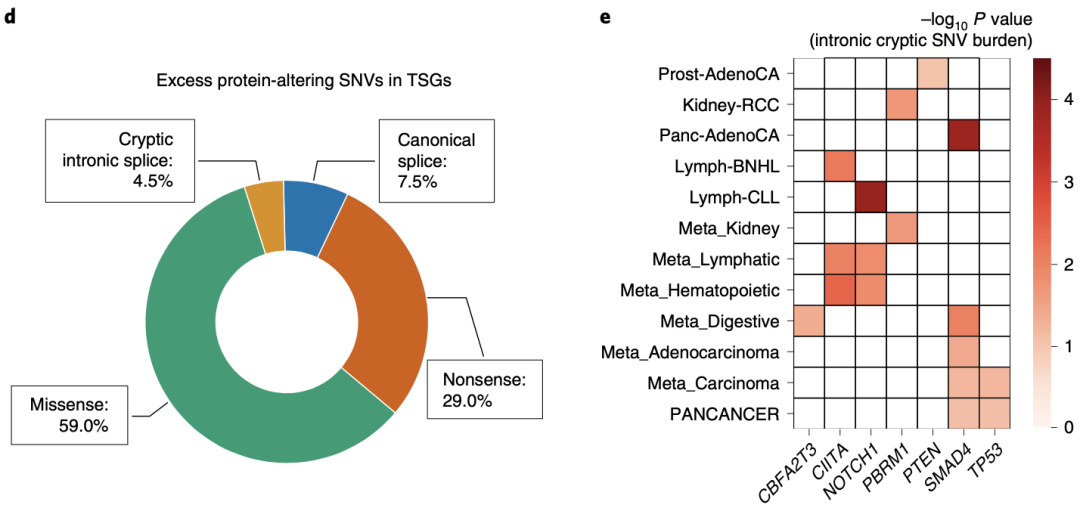
抑癌基因中潜在驱动SNV各类型占比及7个具有显著的内含子隐蔽剪接SNV负担的抑癌基因
有趣的是,对于原癌基因,隐蔽剪接SNV发生的频率并不高,这提示隐蔽剪接SNV应该是导致功能丧失,而不是功能激活。
此外,研究人员还使用Dig算法研究了基因启动子中插入缺失标记(indels)的负荷。结果显示,TP53启动子是唯一具有全基因组显著indels负担的元件,且全为缺失突变(大部分缺失超过4个碱基)。这些突变集中在5’非翻译区,影响转录因子结合,抑制TP53的转录,从而驱动肿瘤发生。
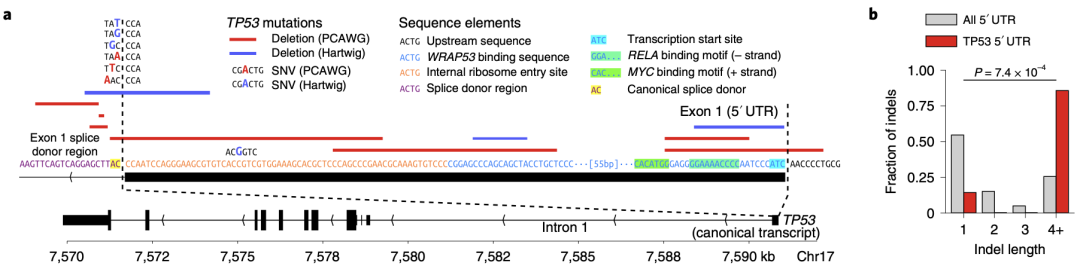
TP53的5’非翻译区发生indels导致肿瘤发生
对106个肿瘤抑制基因和95个具有多外显子5’非翻译区的癌基因的5’非翻译区进行计算发现,ELF3的5’非翻译区同样有显著的SNV负担。然而,因为相应转录数据获取受限,研究人员无法对这些5’非翻译区突变的功能进行进一步分析。
总的来说,研究人员基于概率深度学习方法开发的Dig算法可快速、准确地在测试基因组中寻找潜在的突变基因,其在构建中性突变率模型和识别候选驱动基因方面的强大性能,突出了深度学习在该类研究中的广阔前景。
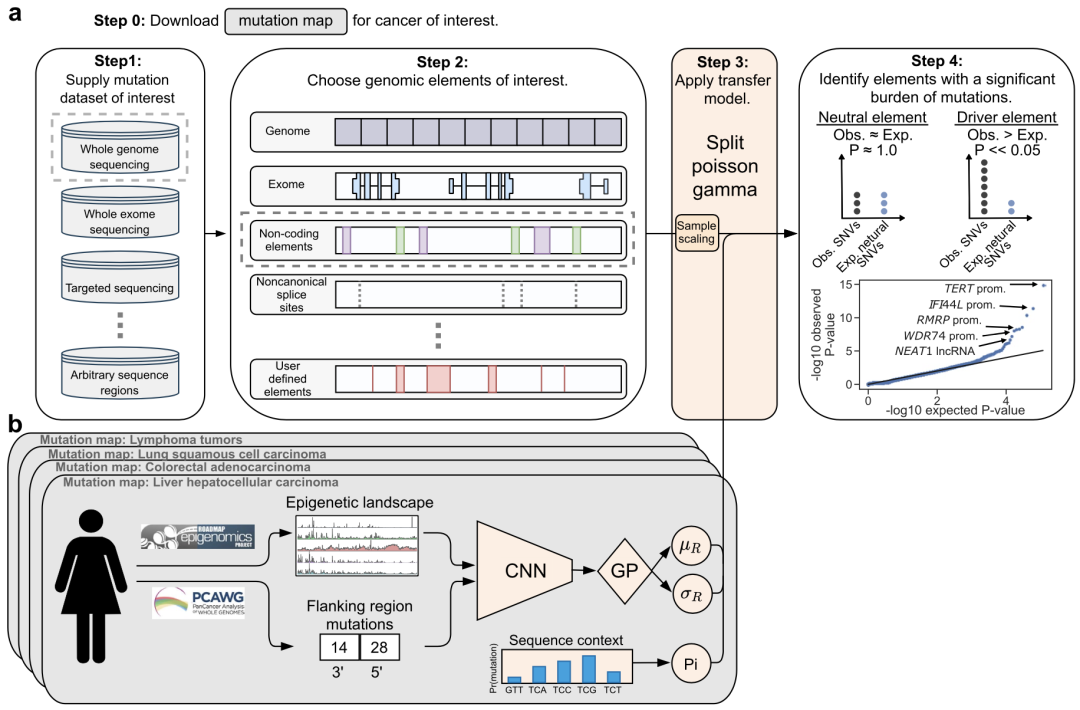
Dig算法概览
由于Dig算法可快速评估分布在大范围基因组上的突变,因此其特别适用于识别弱或中等具有正向选择作用的驱动基因。
然而,该研究也有一定的缺陷,该研究仅靠计算预测,并不足以确定某一突变在肿瘤发生及发展中的因果作用,过量的突变率并不意味着其一定具有正向选择作用。因此,为了确定突变与肿瘤驱动因素的因果作用尚需实验验证。
参考文献
1.Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell 2011, 144(5):646-674.
2.Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence M, Reynolds A, Rynes E, Vlahovicek K, Stamatoyannopoulos JA et al: Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015, 518(7539):360-364.
3.Sherman MA, Yaari AU, Priebe O, Dietlein F, Loh PR, Berger B: Genome-wide mapping of somatic mutation rates uncovers drivers of cancer. Nat Biotechnol 2022.
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
