
Nature methods | 从序列到结构:探索CombFold组装算法的三重奏
来源:生物探索 2024-02-13 11:41
CombFold能够有效地预测出大型蛋白质复合体的三维结构,即使是对于那些由多个不同亚基构成、结构复杂度高的复合体也同样适用。
2月7日发表于Nature Methods的研究“CombFold: predicting structures of large protein assemblies using a combinatorial assembly algorithm and AlphaFold2”,提出了CombFold方法,这是一种结合了组合装配算法(combinatorial assembly algorithm)和AlphaFold2的技术,用于预测大型蛋白质复合体的结构。
在传统的结构表征技术,如X射线晶体学(X-ray crystallography)和核磁共振(Nuclear Magnetic Resonance Spectroscopy)中,大型蛋白质复合体的结构决定尤其具有挑战性。虽然低温电子显微镜(cryo-electron microscopy, cryo-EM)取得了一些进展,但高通量结构测定仍然具有挑战性。CombFold的提出,旨在通过利用AlphaFold2预测的亚基间配对相互作用(pairwise interactions),通过一种层级化和组合式的装配策略,准确预测大型蛋白质复合体的结构。
CombFold的准确度得到了验证,它在两个数据集中预测的复合体中,有72%的复合体在前10个预测中的TM得分(TM-score)超过0.7,表明了其高精度。此外,与Protein Data Bank (PDB)条目相比,预测的复合体结构覆盖率高出20%。CombFold还支持基于交联质谱(crosslinking mass spectrometry)的距离限制和快速枚举可能的复合体化学计量学(complex stoichiometries)。CombFold的高精度使其成为了扩展结构覆盖范围到单体蛋白质以外的有希望的工具。

Highlights
创新的组合装配算法(Combinatorial Assembly Algorithm): CombFold通过结合AlphaFold2的深度学习技术和一种创新的组合装配算法,成功预测了大型蛋白质复合体的结构。这一方法突破了仅依靠传统技术或单一计算模型难以解决的大型蛋白质复合体结构预测的限制。
高精度预测: 在两个数据集中,CombFold能够在前10个预测中以TM得分(TM-score)超过0.7的高精度预测出72%的蛋白质复合体结构,这显示了其在大型蛋白质复合体结构预测方面的高效能。
结构覆盖率提高: 与现有的蛋白质数据库Protein Data Bank (PDB)条目相比,CombFold预测的复合体结构展示了20%的结构覆盖率提高,这意味着能够为科学研究提供更完整的结构信息。
支持距离限制和复合体化学计量学枚举: CombFold不仅能够准确预测结构,还支持基于交联质谱(crosslinking mass spectrometry)的距离限制和快速枚举可能的复合体化学计量学(complex stoichiometries),进一步增强了其在结构生物学研究中的应用潜力。
应用范围广泛: 该方法不仅限于单个蛋白质的结构预测,还能够处理没有已知结构的复杂蛋白质组装体,为复杂生物分子机制的解析和药物开发提供有力的工具。
Strategies
该研究中,CombFold方法的核心是结合AlphaFold2的深度学习能力与一种创新的组合式装配算法(combinatorial assembly algorithm),以预测大型蛋白质复合体的三维结构。这种方法的具体实现步骤如下:
利用AlphaFold2预测亚基间的配对相互作用(Pairwise Interactions): 首先,CombFold使用AlphaFold2对复合体中所有可能的亚基对(subunit pairs)进行结构预测,获取亚基间的相互作用信息。这一步骤是基于AlphaFold2对单链蛋白质或蛋白质复合体的预测能力进行的扩展。
组合和层级化装配(Combinatorial and Hierarchical Assembly): 基于从AlphaFold2获得的亚基间配对相互作用信息,CombFold采用一种组合和层级化的策略来组装蛋白质复合体。这一过程通过优化亚基之间的配对组合,来寻找最可能正确的装配方式。
生成配对子单元的统一表示(Unified Representation of Subunit Pairings): 为了准备进行最终的组装阶段,需要从AlphaFold2预测的多个模型中选择代表性的亚基结构。这一步骤涉及到对每个亚基在不同配对条件下预测的结构进行综合评估,选出最具代表性的结构用于后续的组装。
计算亚基间的转换(Transformations between Subunits): 根据选出的代表性亚基结构,计算亚基间可能的空间位置转换,包括旋转和平移。这些转换信息用于在组装过程中正确定位各个亚基。
组合装配算法(Combinatorial Assembly Algorithm): 在得到所有必要的亚基间转换信息后,CombFold采用组合装配算法对亚基进行层级化组装。这一算法通过枚举所有可能的亚基组合和相应的空间定位,寻找最终能够正确组装出整个复合体的方法。
距离限制和化学计量学枚举(Distance Restraints and Stoichiometry Enumeration): CombFold支持基于交联质谱数据的距离限制,以及对未知结构的复合体进行化学计量学(stoichiometry)的快速枚举,这一功能使得CombFold能够在缺乏完整结构信息的情况下,也能进行高度可信的结构预测。
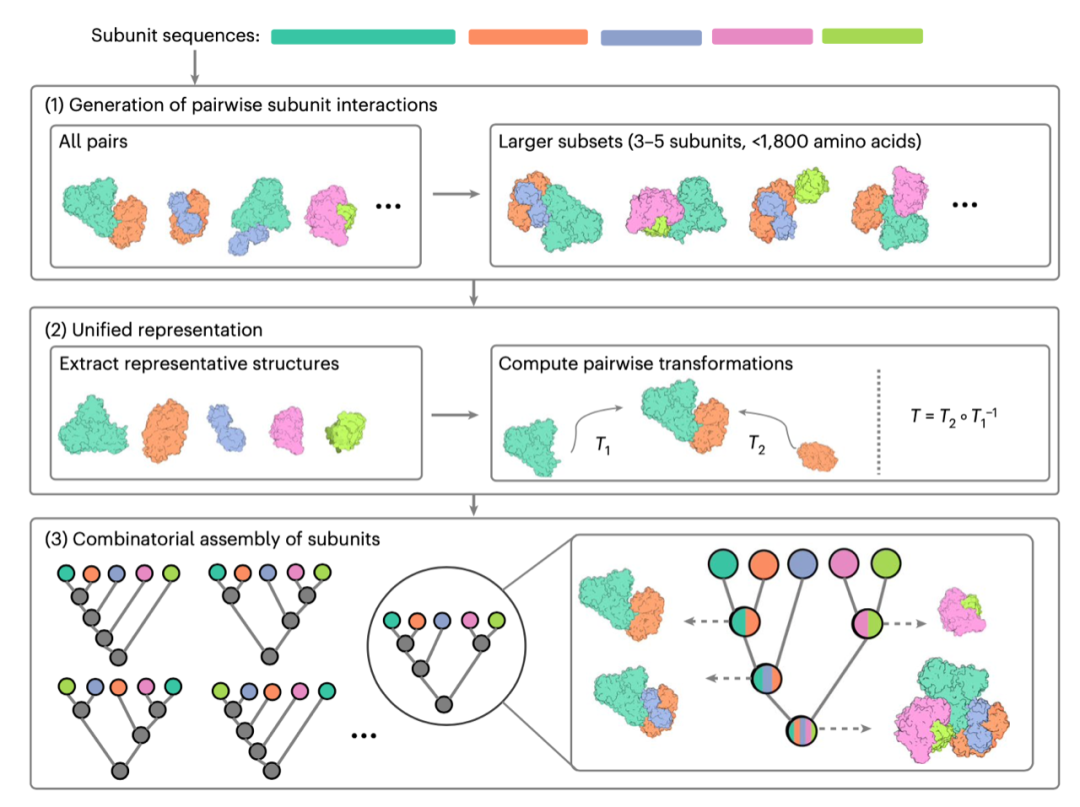
CombFold的三个阶段(Credit: Nature methods)
Prospects
动态结构预测: 当前CombFold主要预测蛋白质复合体的静态结构。然而,许多蛋白质在生物学过程中会呈现出动态变化和多种构象。如何预测和理解这些动态过程和构象变化仍是一个挑战。
复合体中未知亚基的预测: 对于一些复合体,其组成亚基的完整信息可能未知。当前的方法需要已知复合体的化学计量学(stoichiometry)信息,对于那些部分亚基未知或存在亚基变异的复合体,如何有效预测它们的结构尚未明确。
大规模复合体的处理能力: 对于极其庞大的蛋白质复合体,即使是CombFold这样的先进方法也可能面临计算资源和算法效率的限制。如何提高算法的处理能力,以应对更大规模的复合体预测,是一个待解决的问题。
复合体异质性的解析: 许多蛋白质复合体在生物体内以不同的亚基组合形式存在,展现出高度的异质性。如何准确捕捉并预测这种复合体的多样性和异质性是一个重要的研究方向。
实验数据的整合和利用: CombFold虽然能够整合交联质谱(crosslinking mass spectrometry)等实验数据,但如何更有效地利用其他类型的实验数据(如FRET、Cryo-EM等),以提高预测精度和可靠性,还需要进一步探索。
预测结果的验证: CombFold预测的结构需要通过实验方法进行验证。如何系统地对预测结果进行验证,并根据验证结果优化算法,是实现预测方法实用化的关键一步。
应用领域的扩展: CombFold的潜在应用领域非常广泛,如何在药物设计、疾病机理研究等领域具体应用这些预测技术,以及如何根据应用需求进一步优化算法,都是未来研究的重要内容。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
