
全文编译!Cell子刊指出新冠肺炎其实是由新型冠状病毒引发的一种古老的细胞因子释放综合征
来源:本站原创 2020-04-26 16:21
2020年4月26日讯/生物谷BIOON/---近二十年来,严重急性呼吸综合征(SARS)冠状病毒(SARS-CoV)和中东呼吸综合征(MERS)冠状病毒(MERS-CoV)从动物传播给人类,在流行地区分别引起严重呼吸道疾病:SARS和MERS。2019年12月,在一些传染性呼吸道疾病患者中发现的另一种称为SARS-CoV-2的新型冠状病毒具有人与人之间的传
2020年4月26日讯/生物谷BIOON/---近二十年来,严重急性呼吸综合征(SARS)冠状病毒(SARS-CoV)和中东呼吸综合征(MERS)冠状病毒(MERS-CoV)从动物传播给人类,在流行地区分别引起严重呼吸道疾病:SARS和MERS。2019年12月,在一些传染性呼吸道疾病患者中发现的另一种称为SARS-CoV-2的新型冠状病毒具有人与人之间的传播能力。这种疾病目前被称为2019年冠状病毒病(COVID-19,也称为新冠肺炎)。这种新型冠状病毒已在全球范围内迅速传播,导致疫情大爆发。COVID-19由致病性的SARS-CoV-2诱发,截至2020年4月9日,共有1484811例病例,死亡88538例。COVID-19的主要表型是严重急性呼吸窘迫综合征(ARDS),就像SARS-CoV和MERS-CoV引起的疾病一样。
Lu等人和Zhou等人报告了SARS-CoV-2的基因组学特征和流行病学。SARS-CoV2的基因组序列与SARS-CoV和MERS-CoV相似,但又有区别,这是因为它与SARS-CoV基因组的序列一致性(sequence identity)大约为80%,与MERS-CoV基因组的序列一致性大约为50%。令人关注的是,SARS-CoV-2与中国东部地区采集的两种蝙蝠冠状病毒--- bat-SL-CoVZC45和bat-SL-CoVZXC21---在全基因组水平上的一致性大约为90%。蛋白序列分析表明,SARS-CoV-2与SARS-CoV一样,具有7个保守的非结构域,这表明这两种冠状病毒有一定的相关性。此外,SARS-CoV-2的受体结合结构域与SARS-CoV相似,尽管在一些关键残基上存在着氨基酸变化。因此,SARS-CoV-2有可能使用与SARS-CoV相同的细胞进入受体---血管紧张素转换酶II(ACE2)。
考虑到COVID-19的高死亡率(在全球为5.96%),开发有效的治疗方法是一个迫切的问题,这需要确定高质量的靶点。为此,有两篇论文确定了ACE2为SARS-CoV-2的细胞进入受体。此外,Hoffman及其同事们发现受体介导的病毒进入依赖于一种丝氨酸蛋白酶---跨膜丝氨酸蛋白酶2(TMPRSS2)。值得注意的是,临床上批准的TMPRSS2抑制剂可以阻止SARS-CoV-2进入细胞。鉴于肺泡2型细胞(ATII)在稳态下高度表达ACE2和TMPRSS2,这些细胞可能是SARS-CoV-2在肺部中的主要进入细胞。
Zhou等人利用表达或不表达来自人类、中华马蹄蝠、果子狸、猪和小鼠的ACE2蛋白的HeLa细胞进行了病毒感染性研究。他们发现SARS-CoV-2可以进入表达除小鼠ACE2蛋白之外的其他ACE2蛋白的细胞,但不能进入缺乏ACE2蛋白的细胞,这表明这种病毒利用ACE2作为它的进入受体。此外,SARS-CoV-2不能进入缺乏ACE2但表达二肽基肽酶4(DPP4)或氨基肽酶N(APN)的细胞,其中DPP4和APN分别是MERS-CoV和HCoV-229E的进入受体。
众所周知,冠状病毒进入细胞需要病毒刺突蛋白(S)的S1亚基与细胞表面受体结合,然后S蛋白的S2亚基介导病毒与细胞膜融合。这个过程需要宿主细胞蛋白酶对S蛋白进行激活,使得在S1/S2和S2'位点对S蛋白进行切割。分析哪些细胞因子被SARS-CoV-2用于细胞进入,应当能够提供对这种病毒传播的新见解,并揭示有效的治疗靶点。
Hoffmann等对SARS-CoV-2的细胞进入机制进行了详细分析。他们证实SARS-CoV-2利用ACE2进入细胞,TMPRSS2和内体半胱氨酸蛋白酶cathepsin B和L(CatB/L)用于S蛋白激活。他们还发现,SARS-CoV-2 S蛋白(SARS2-S)的S1/S2位点在293T细胞中被有效地切割。此外,他们利用携带SARS-2-S或SARS-CoV S蛋白(SARS-S)的复制缺陷性的水泡性口炎病毒(VSV)颗粒开展实验,他们发现不论是SARS-2-S还是SARS-S,它们能够进入的一系列来自不同动物的细胞系都是相同的,这与SARS-S和ACE2结合所必需的氨基酸残基在SARS-2-S中也是保守的发现相一致。事实上,当迫使BHK-21细胞等ACE2阴性细胞表达人ACE2或蝙蝠ACE2时,SARS2-S和 SARS-S都能进入这些细胞,但是不能进入表达人DPP4或APN的ACE2阴性细胞。再者,针对人ACE2的抗体阻断了SARS-S-S和SARS-2-S进入人细胞系。这些结果表明,SARS-2-S与SARS-S一样都是利用ACE2进入细胞。
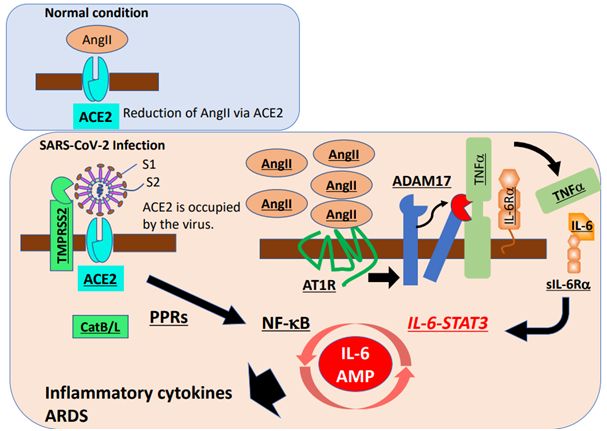
Hoffmann等人随后研究了SARS-CoV-2进入细胞的蛋白酶依赖性。SARS-2-S驱动的293T细胞(表达ACE2,但不表达TIMPRSS2)进入可被CatB/L的抑制剂氯化铵抑制,然而这种抑制剂对SARS-2-S驱动的Caco-2细胞(表达TIMPRSS2)进入的抑制效率较低。经临床验证的TMPRSS2抑制剂---甲磺酸卡莫司他(camostat mesylate)---部分抑制了SARS-2-S驱动的Caco-2细胞进入,而甲磺酸卡莫司他与CatB/L抑制剂E-64d一起完全抑制了SARS-CoV-2进入,这表明TIMPRSS2和CatB/L都参与了SARS-CoV-2进入。然而,TMPRSS2的强行表达可确保SARS-2-S依赖性的293T细胞(CatB/L受到抑制)进入,这表明当细胞表达TMPRSS2时,无论CatB/L是否表达,这都可诱导SARS-CoV-2进入细胞。SARS-2-S驱动的肺细胞进入也被甲磺酸卡莫司他所抑制。这些研究结果表明,SARS-CoV-2进入细胞依赖于ACE2和TMPRSS2等表面分子。
Zhou等人和Hoffmann等人一起研究发现,SARS-CoV-2与SARS-CoV一样,利用ACE2作为细胞进入受体。重要的是,这些结果提示了针对COVID19的治疗靶点。一个靶点是SARS-2-S蛋白与ACE2之间的结合,另一个靶点是可激活SARS-2-S蛋白的TMPRSS的丝氨酸蛋白酶活性。
这些治疗靶点可能对SARS-CoV-2感染的初始阶段起作用,但对这种疾病的后期阶段不占优势,这是因为后期阶段诱导的极强慢性炎症是ARDS介导的死亡的主要原因。一直以来,ARDS是一种由肺炎、败血症或因“细胞因子风暴(cytokine storm)”引起的肺吸入导致的致命综合征,其中在细胞因子风暴中,免疫细胞和非免疫细胞会释放大量的促炎性细胞因子,从而对宿主造成损伤。NF-κB途径的过度激活参与了这种表型。在冠状病毒感染后,NF-κB激活的主要途径之一是基于模式识别受体(PRR)的MyD88途径,从而诱导多种促炎性细胞因子,包括IL-6、TNFα和趋化因子。
ACE2是一种膜蛋白,也是血管紧张素2(AngII)的灭活剂。重要的是,ACE2与SARS-CoV一起被内吞,导致细胞表面上的ACE2减少,随后便是血清AngII增加。鉴于ACE2在肺损伤模型中也被下调,而且重组ACE2可抑制ARDS的发生,因此严重的肺部炎症本身可能会诱导肾素-血管紧张素(renin-angiotensin)途径的失调,继而在SARS-CoV-2感染后出现ARDS。事实上,在动物模型中,SARS-CoV诱导的ARDS可以通过血管紧张素受体1型(AT1R)的抑制剂来预防。
AngII不仅作为血管收缩剂,而且还通过AT1R作为促炎性细胞因子发挥作用。AngII-AT1R轴还能激活NF-κB、解离素(disintegrin)和金属蛋白酶17(ADAM17),从而产生两种NF-κB激活物:成熟型的表皮生长因子受体(EGFR)配体和TNFα。ADAM17诱导还可将IL-6Rα的膜结合形式加工成可溶性形式(sIL-6Rα),随后在各种IL-6Rα阴性的非免疫细胞(包括成纤维细胞、内皮细胞和上皮细胞)中,通过sIL-6Rα-IL-6复合物进行gp130介导的STAT3激活。STAT3是充分激活NF-κB途径所必需的,而STAT3在体内的主要刺激因子是IL-6,尤其是在炎症期间,尽管至少在体外IL-6家族细胞因子的9种其他成员也可以激活STAT3。因此,呼吸系统中的SARS-CoV-2感染可同时激活NF-κB和STAT3,进而激活IL-6放大器(IL-6 amplifier, IL-6 Amp),这是STAT3对NF-κB的超激活机制,可导致多种炎症和自身免疫疾病的发生。IL-6 Amp诱导包括IL-6在内的多种促炎性细胞因子和趋化因子,并在病变部位招募淋巴细胞和髓系细胞,比如活化的T细胞和巨噬细胞,从而在一种正反馈循环中强化IL-6 Amp(图1)。重要的是,由于IL-6是细胞衰老的主要功能标志物,IL-6 Amp的年龄依赖性增强可能与COVID-19死亡率的年龄依赖性增加相对应。
事实上,在SARS-CoV-2感染时观察到的ARDS是细胞因子释放综合征(CRS),这是一种由细胞因子风暴诱发的疾病。用嵌合抗原受体(CAR)-T细胞疗法治疗白血病和淋巴瘤时发现的CRS的致命副作用也与炎性细胞因子升高有关。这些升高的促炎性细胞因子可能是由IL-6 Amp诱导的。考虑到抗IL-6R抗体托珠单抗(tocilizumab)是CAR-T细胞疗法中治疗CRS的有效药物,科学家们可能考虑针对COVID-19中出现的CRS使用具有类似作用机制的药物。
综上所述,所有证据都支持这样的观点,即在COVID-19中观察到的ARDS是由AngII-AT1R轴和PRR通路以及IL-6-STAT3轴诱导的CRS,这提示着IL-6/gp130信号通路可能是治疗COVID-19的治疗性靶点。(生物谷 Bioon.com)
参考资料:
Toshio Hirano et al. COVID-19: a new virus, but an old cytokine release syndrome. Immunity, 2020, doi:10.1016/j.immuni.2020.04.003.
Lu等人和Zhou等人报告了SARS-CoV-2的基因组学特征和流行病学。SARS-CoV2的基因组序列与SARS-CoV和MERS-CoV相似,但又有区别,这是因为它与SARS-CoV基因组的序列一致性(sequence identity)大约为80%,与MERS-CoV基因组的序列一致性大约为50%。令人关注的是,SARS-CoV-2与中国东部地区采集的两种蝙蝠冠状病毒--- bat-SL-CoVZC45和bat-SL-CoVZXC21---在全基因组水平上的一致性大约为90%。蛋白序列分析表明,SARS-CoV-2与SARS-CoV一样,具有7个保守的非结构域,这表明这两种冠状病毒有一定的相关性。此外,SARS-CoV-2的受体结合结构域与SARS-CoV相似,尽管在一些关键残基上存在着氨基酸变化。因此,SARS-CoV-2有可能使用与SARS-CoV相同的细胞进入受体---血管紧张素转换酶II(ACE2)。
考虑到COVID-19的高死亡率(在全球为5.96%),开发有效的治疗方法是一个迫切的问题,这需要确定高质量的靶点。为此,有两篇论文确定了ACE2为SARS-CoV-2的细胞进入受体。此外,Hoffman及其同事们发现受体介导的病毒进入依赖于一种丝氨酸蛋白酶---跨膜丝氨酸蛋白酶2(TMPRSS2)。值得注意的是,临床上批准的TMPRSS2抑制剂可以阻止SARS-CoV-2进入细胞。鉴于肺泡2型细胞(ATII)在稳态下高度表达ACE2和TMPRSS2,这些细胞可能是SARS-CoV-2在肺部中的主要进入细胞。
Zhou等人利用表达或不表达来自人类、中华马蹄蝠、果子狸、猪和小鼠的ACE2蛋白的HeLa细胞进行了病毒感染性研究。他们发现SARS-CoV-2可以进入表达除小鼠ACE2蛋白之外的其他ACE2蛋白的细胞,但不能进入缺乏ACE2蛋白的细胞,这表明这种病毒利用ACE2作为它的进入受体。此外,SARS-CoV-2不能进入缺乏ACE2但表达二肽基肽酶4(DPP4)或氨基肽酶N(APN)的细胞,其中DPP4和APN分别是MERS-CoV和HCoV-229E的进入受体。
众所周知,冠状病毒进入细胞需要病毒刺突蛋白(S)的S1亚基与细胞表面受体结合,然后S蛋白的S2亚基介导病毒与细胞膜融合。这个过程需要宿主细胞蛋白酶对S蛋白进行激活,使得在S1/S2和S2'位点对S蛋白进行切割。分析哪些细胞因子被SARS-CoV-2用于细胞进入,应当能够提供对这种病毒传播的新见解,并揭示有效的治疗靶点。
Hoffmann等对SARS-CoV-2的细胞进入机制进行了详细分析。他们证实SARS-CoV-2利用ACE2进入细胞,TMPRSS2和内体半胱氨酸蛋白酶cathepsin B和L(CatB/L)用于S蛋白激活。他们还发现,SARS-CoV-2 S蛋白(SARS2-S)的S1/S2位点在293T细胞中被有效地切割。此外,他们利用携带SARS-2-S或SARS-CoV S蛋白(SARS-S)的复制缺陷性的水泡性口炎病毒(VSV)颗粒开展实验,他们发现不论是SARS-2-S还是SARS-S,它们能够进入的一系列来自不同动物的细胞系都是相同的,这与SARS-S和ACE2结合所必需的氨基酸残基在SARS-2-S中也是保守的发现相一致。事实上,当迫使BHK-21细胞等ACE2阴性细胞表达人ACE2或蝙蝠ACE2时,SARS2-S和 SARS-S都能进入这些细胞,但是不能进入表达人DPP4或APN的ACE2阴性细胞。再者,针对人ACE2的抗体阻断了SARS-S-S和SARS-2-S进入人细胞系。这些结果表明,SARS-2-S与SARS-S一样都是利用ACE2进入细胞。
图1.细胞因子释放综合征COVID-19的潜在治疗靶点。图片来自Immunity, 2020, doi:10.1016/j.immuni.2020.04.003。
Hoffmann等人随后研究了SARS-CoV-2进入细胞的蛋白酶依赖性。SARS-2-S驱动的293T细胞(表达ACE2,但不表达TIMPRSS2)进入可被CatB/L的抑制剂氯化铵抑制,然而这种抑制剂对SARS-2-S驱动的Caco-2细胞(表达TIMPRSS2)进入的抑制效率较低。经临床验证的TMPRSS2抑制剂---甲磺酸卡莫司他(camostat mesylate)---部分抑制了SARS-2-S驱动的Caco-2细胞进入,而甲磺酸卡莫司他与CatB/L抑制剂E-64d一起完全抑制了SARS-CoV-2进入,这表明TIMPRSS2和CatB/L都参与了SARS-CoV-2进入。然而,TMPRSS2的强行表达可确保SARS-2-S依赖性的293T细胞(CatB/L受到抑制)进入,这表明当细胞表达TMPRSS2时,无论CatB/L是否表达,这都可诱导SARS-CoV-2进入细胞。SARS-2-S驱动的肺细胞进入也被甲磺酸卡莫司他所抑制。这些研究结果表明,SARS-CoV-2进入细胞依赖于ACE2和TMPRSS2等表面分子。
Zhou等人和Hoffmann等人一起研究发现,SARS-CoV-2与SARS-CoV一样,利用ACE2作为细胞进入受体。重要的是,这些结果提示了针对COVID19的治疗靶点。一个靶点是SARS-2-S蛋白与ACE2之间的结合,另一个靶点是可激活SARS-2-S蛋白的TMPRSS的丝氨酸蛋白酶活性。
这些治疗靶点可能对SARS-CoV-2感染的初始阶段起作用,但对这种疾病的后期阶段不占优势,这是因为后期阶段诱导的极强慢性炎症是ARDS介导的死亡的主要原因。一直以来,ARDS是一种由肺炎、败血症或因“细胞因子风暴(cytokine storm)”引起的肺吸入导致的致命综合征,其中在细胞因子风暴中,免疫细胞和非免疫细胞会释放大量的促炎性细胞因子,从而对宿主造成损伤。NF-κB途径的过度激活参与了这种表型。在冠状病毒感染后,NF-κB激活的主要途径之一是基于模式识别受体(PRR)的MyD88途径,从而诱导多种促炎性细胞因子,包括IL-6、TNFα和趋化因子。
ACE2是一种膜蛋白,也是血管紧张素2(AngII)的灭活剂。重要的是,ACE2与SARS-CoV一起被内吞,导致细胞表面上的ACE2减少,随后便是血清AngII增加。鉴于ACE2在肺损伤模型中也被下调,而且重组ACE2可抑制ARDS的发生,因此严重的肺部炎症本身可能会诱导肾素-血管紧张素(renin-angiotensin)途径的失调,继而在SARS-CoV-2感染后出现ARDS。事实上,在动物模型中,SARS-CoV诱导的ARDS可以通过血管紧张素受体1型(AT1R)的抑制剂来预防。
AngII不仅作为血管收缩剂,而且还通过AT1R作为促炎性细胞因子发挥作用。AngII-AT1R轴还能激活NF-κB、解离素(disintegrin)和金属蛋白酶17(ADAM17),从而产生两种NF-κB激活物:成熟型的表皮生长因子受体(EGFR)配体和TNFα。ADAM17诱导还可将IL-6Rα的膜结合形式加工成可溶性形式(sIL-6Rα),随后在各种IL-6Rα阴性的非免疫细胞(包括成纤维细胞、内皮细胞和上皮细胞)中,通过sIL-6Rα-IL-6复合物进行gp130介导的STAT3激活。STAT3是充分激活NF-κB途径所必需的,而STAT3在体内的主要刺激因子是IL-6,尤其是在炎症期间,尽管至少在体外IL-6家族细胞因子的9种其他成员也可以激活STAT3。因此,呼吸系统中的SARS-CoV-2感染可同时激活NF-κB和STAT3,进而激活IL-6放大器(IL-6 amplifier, IL-6 Amp),这是STAT3对NF-κB的超激活机制,可导致多种炎症和自身免疫疾病的发生。IL-6 Amp诱导包括IL-6在内的多种促炎性细胞因子和趋化因子,并在病变部位招募淋巴细胞和髓系细胞,比如活化的T细胞和巨噬细胞,从而在一种正反馈循环中强化IL-6 Amp(图1)。重要的是,由于IL-6是细胞衰老的主要功能标志物,IL-6 Amp的年龄依赖性增强可能与COVID-19死亡率的年龄依赖性增加相对应。
事实上,在SARS-CoV-2感染时观察到的ARDS是细胞因子释放综合征(CRS),这是一种由细胞因子风暴诱发的疾病。用嵌合抗原受体(CAR)-T细胞疗法治疗白血病和淋巴瘤时发现的CRS的致命副作用也与炎性细胞因子升高有关。这些升高的促炎性细胞因子可能是由IL-6 Amp诱导的。考虑到抗IL-6R抗体托珠单抗(tocilizumab)是CAR-T细胞疗法中治疗CRS的有效药物,科学家们可能考虑针对COVID-19中出现的CRS使用具有类似作用机制的药物。
综上所述,所有证据都支持这样的观点,即在COVID-19中观察到的ARDS是由AngII-AT1R轴和PRR通路以及IL-6-STAT3轴诱导的CRS,这提示着IL-6/gp130信号通路可能是治疗COVID-19的治疗性靶点。(生物谷 Bioon.com)
参考资料:
Toshio Hirano et al. COVID-19: a new virus, but an old cytokine release syndrome. Immunity, 2020, doi:10.1016/j.immuni.2020.04.003.
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
87%用户都在用生物谷APP 随时阅读、评论、分享交流 请扫描二维码下载->
