
NEJM:世界首次!在子宫内成功治疗罕见遗传病
来源:生物世界 2025-02-26 16:02
该研究记录了首例尚在子宫中的胎儿接受利司扑兰治疗的案例,标志着神经肌肉退行性疾病的子宫内干预的里程碑,若疗效及安全性获进一步验证,或将改写 SMA 的临床管理范式。
这个患有严重的 1 型脊髓性肌萎缩症(SMA-1)的小女孩,没有表现出任何运动神经元疾病的迹象,已经快三岁的她,已远超 SMA-1 患者仅 8.5 个月的中位生存期。
而这一切,源于她尚在母亲子宫中就接受了一种基因靶向药物的治疗,她也是第一个在子宫内接受罕见遗传病治疗的人,出生后,她仍在继续用药,如今,她已出生 30 个月,她遗传的致病运动神经元疾病得到了有效治疗。
2025年2月19日,国际顶尖医学期刊《新英格兰医学杂志》(NEJM)报道了这一治疗研究。

脊髓性肌萎缩症(SMA)是一种毁灭性的运动神经元疾病,其主要特征是运动神经元死亡、肌肉失神经支配和肌肉无力,患者通常在 2 岁前因呼吸衰竭而死亡。该疾病由 SMN1 基因功能缺失突变,导致无法产生足够的 SMN 蛋白,而 SMN2 基因也能产生少量 SMN 蛋白,SMN2 基因拷贝数与疾病的严重程度负相关,SMN2 仅为 1 个或 2 个拷贝时,患者表现为严重的 1 型脊髓性肌萎缩症(SMA-1),此类患者的中位生存期仅 8.5 个月。
这名小女孩尚在母亲腹中时,因存在患有严重的 1 型脊髓性肌萎缩症(SMA-1)风险而接受羊膜穿刺术进行检测,她此前已有一名兄长(已故)经基因检测确诊为 SMA-1。检测结果显示,胎儿的 SMN1 基因零拷贝(确诊患有 SMA),且 SMN2 基因仅两个拷贝(拷贝数越少越严重),这表明其患有严重的 SMA-1。
利司扑兰(Risdiplam)是罗氏公司开发的一种调控 SMN2 基因剪接的小分子口服药物,可提高脊髓性肌萎缩症(SMA)患者的 SMN 蛋白水平,从而改善疾病表现。动物模型研究显示,利司扑兰能够突破胎盘屏障,在胎儿中枢神经系统达到治疗浓度,这支持了其用于产前治疗的可行性。 SMA 的运动神经元变性始于妊娠 18-24 周,在此之前治疗可能更大程度地挽救尚未凋亡的运动神经元。
基因诊断确诊后,胎儿的父母建议医生在孩子出生前就开始治疗,因为他们此前已经因为这种疾病失去了一个孩子。针对他们的情况,FDA 批准了这种子宫内治疗。这位母亲在妊娠 32 周时开始连续 6 周每天服用利司扑兰,而这名婴儿在出生后第 8 天开始每天服用利司扑兰至今,截至 2025 年 2 月,她已出生 30 个月,这也远超 SMA-1 患者的中位生存期,而她目前没有表现出任何运动神经元疾病的迹象。
检测结果显示,治疗期间,母体血液中药物浓度为 14 ng/mL,脐带血中浓度为 9.7 ng/mL。羊水中浓度为 4.6 ng/mL,这表明利司扑兰确实可以通过胎盘,但胎儿暴露量低于母体。婴儿血液中 SMN 蛋白水平显著高于基线水平,婴儿血浆中磷酸化重链神经丝蛋白(pNfH)水平下降,说明神经元损伤减轻。
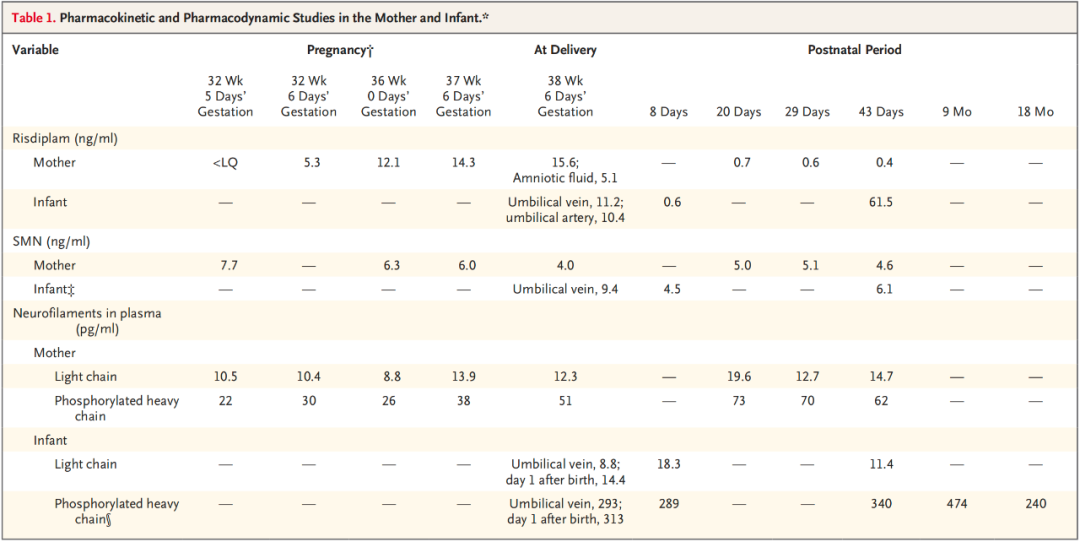
在出生后 30 个月时,未出现肌张力低下、反射消失等典型的 SMA 症状,且神经肌肉发育正常。此外,患者出现了双侧视神经发育不良伴轻度近视,左中脑发育不良致右侧轻偏瘫以及全面性发育迟缓。研究团队认为这些发育异常是因为治疗时已经出现了部分发育问题,与治疗药物无关。因此,未来可进一步探索孕早期治疗能否进一步预防发育异常。
该研究记录了首例尚在子宫中的胎儿接受利司扑兰治疗的案例,标志着神经肌肉退行性疾病的子宫内干预的里程碑,若疗效及安全性获进一步验证,或将改写 SMA 的临床管理范式。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
