
轰动学界的Nature和Cell论文,再被质疑有误,Ptbp1缺失不能诱导胶质细胞转化为神经元
来源:生物世界 2023-06-09 16:31
补充因疾病而失去的细胞是再生医学领域的“圣杯”,然而,成年哺乳动物(包括人类)的中枢神经系统(CNS)在很大程度上已经失去了这种再生能力。
补充因疾病而失去的细胞是再生医学领域的“圣杯”,然而,成年哺乳动物(包括人类)的中枢神经系统(CNS)在很大程度上已经失去了这种再生能力。近年来,有一种新兴策略通过将体内的胶质细胞转化为神经元,这为治疗神经退行性疾病带来了巨大希望。但想实现这一点,不但复杂而且效率低下。
2020年,两篇分别发表于 Cell 和 Nature 的顶刊论文显示, 只需敲低Ptbp1,就能高效诱导胶质细胞向神经元的转化,挽救帕金森病小鼠模型的运动障碍。这为通过胶质细胞向神经元的转分化来治疗神经退行性疾病带来了巨大前景。然而,其他团队并没有能够重复出这两篇论文的结果,人们开始质疑,敲低Ptbp1后,胶质细胞向神经元的转化真的发生了吗?
2020年4月,中科院神经所杨辉团队在 Cell 期刊发表了题为:Glia-to-Neuron Conversion by CRISPR-CasRx Alleviates Symptoms of Neurological Disease in Mice 的论文【1】。该研究发现,使用CRISPR-CasRx系统敲低Ptbp1,能够诱导胶质细胞转化为神经元。
2020年6月,加州大学圣地亚哥分校付向东团队在 Nature 发表了题为:Reversing a model of Parkinson's disease with in situconverted nigral neurons 的论文【2】。该研究发现,使用shRNA或反义寡核苷酸(ASO)敲低Ptbp1蛋白表达,能够诱导胶质细胞转化为神经元。
这两项研究提示了我们,Ptbp1基因可能是治疗包括帕金森病在内的因神经元损伤导致的神经退行性疾病的潜在靶点。
这两篇论文发表后,在业内引起广泛关注和争议。
2021年9月,德州大学西南医学中心张春立团队在 Cell 发表了题为:Revisiting astrocyte to neuron conversion with lineage tracing in vivo 的论文【3】。该研究利用谱系追踪证明,无论是过表达NeuroD1,还是敲低Ptbp1,都不能在体内将胶质细胞转化为神经元,之前的研究中观察到的转分化而来的神经元实际上是大脑中原本就存在的内源性神经元。
2021年10月,约翰·霍普金斯大学医学院 Seth Blackshaw 团队在预印本 bioRxiv 发表了题为:Ptbp1 deletion does not induce glia-to-neuron conversion in adult mouse retina and brain 的论文【4】。该论文指出,在小鼠的视网膜和大脑中,Ptbp1的特异性缺失,都不会诱导胶质细胞向神经元的转化。2022年6月,Seth Blackshaw 团队将部分试验结果发表在了 Cell Reports 期刊【5】。
值得一提的是,2023年2月,杨辉团队在 Gene Therapy 期刊发表了题为:Ptbp1 knockdown failed to induce astrocytes to neurons in vivo 的论文【6】。该研究指出,Cas13x介导的Ptbp1的敲低在体内不能诱导胶质细胞向神经元的转化。从而否定了此前在 Cell 发表的结果,认为之前的结果是由于GFAP-AAV泄漏所致。
2023年6月7日,约翰·霍普金斯大学医学院 Seth Blackshaw 团队在 Nature 发表了题为:Ptbp1 deletion does not induce astrocyte-toneuron conversion 的文章【7】。
这项研究显示,Ptbp1功能缺失并不会诱导星形胶质细胞向神经元的分化,甚至并没有引起星型胶质细胞基因表达的实质性改变。

Seth Blackshaw 团队指出,付向东团队的这篇 Nature 论文中观察到的Ptbp1敲低所诱导的胶质细胞向神经元的转分化的结论缺乏一些必要的直接证据。通过对携带Ptbp1杂合或纯合突变的成熟星形胶质细胞的遗传谱系和单细胞RNA测序(scRNA-Seq)分析显示,Ptbp1缺失的星形胶质细胞并没有向神经元转化,也没有对其他基因表达产生实质性影响。
为了从基因上破坏星形胶质细胞中Ptbp1的功能,研究团队使用了星形胶质细胞特异性他莫昔芬诱导的Aldh1l1creERT2小鼠以及Sun1-GFPloxP/loxP和Ptbp1loxP/loxP细胞系,并使用4-羟基他莫昔芬(4-OHT)诱导星形胶质细胞产生杂合性或纯合型Ptbp1缺失,这些星形胶质细胞被SUN1-GFP不可逆标记用于谱系追踪。在4-OHT诱导后第2、4、8周,在任何基因型小鼠中都没有观察到大脑中新型胶质细胞向神经元的转化。
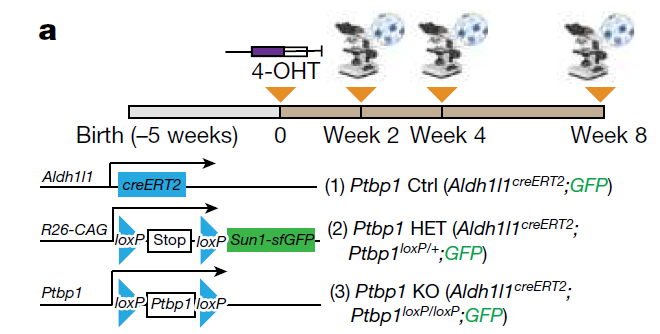
付向东团队的 Nature 论文显示,敲低Ptbp1诱导的胶质细胞转化而来的神经元可以分泌多巴胺,恢复6-羟多巴胺(6-OHDA)损伤多巴胺能神经元诱导的帕金森病小鼠模型的运动功能。
然而,这篇发表在 Nature 的质疑文章显示,6-OHDA损伤的Ptbp1杂合缺失或纯合缺失的小鼠都没有出现新生的神经元。因此,研究团队得出结论:无论是否有神经元损伤,Ptbp1功能缺失都不会导致星形胶质细胞向神经元的转化。
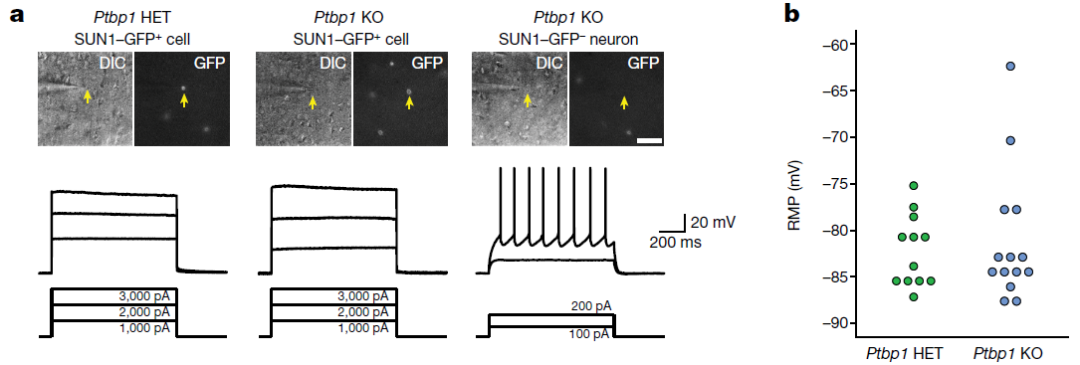
接下来,研究团队检查了Ptbp1缺失是否改变了星形胶质细胞的生理活动。膜片钳实验显示,Ptbp1杂合缺失或纯合缺失都不会诱导星形胶质细胞出现神经元样电生理特性。
为了全面分析Ptbp1缺失在新型胶质细胞中的分子效应,研究团队对4-OHT诱导2周后的野生型、杂合型和纯合型Ptbp1缺失小鼠的整个皮质、纹状体和黑质进行了单细胞RNA测序(scRNA-seq)分析。结果显示,Ptbp1缺失后,大多数星形胶质细胞标志物的表达没有变化,且未观察到神经祖细胞或成熟神经元特异性基因的诱导。免疫染色实验证实Ptbp1缺失的星形胶质细胞保留了胶质细胞标志物SOX9的表达。研究团队得出结论:Ptbp1不会抑制星形胶质细胞中神经元基因的表达。
为了进一步研究Ptbp1功能缺失在稍后时间点诱导的基因表达变化,我们研究团队在4- OHT诱导野生型和纯合Ptbp1缺失小鼠4周后,对GFP阳性星形胶质细胞和荧光活化细胞分选富集的星形胶质细胞来源的细胞进行了进一步的scRNA-seq分析。结果显示,在Ptbp1缺失后,所有三个脑区中神经源性或其他细胞类型特异性标志基因的表达没有差异。大多数星形胶质细胞标志物的表达未发生变化,未观察到神经祖细胞或成熟神经元特异性基因的诱导。这些结果表明,Ptbp1缺失后的星形胶质细胞,没有出现胶质细胞特异性基因的表达,也没有星形胶质细胞特异性基因的表达下调。
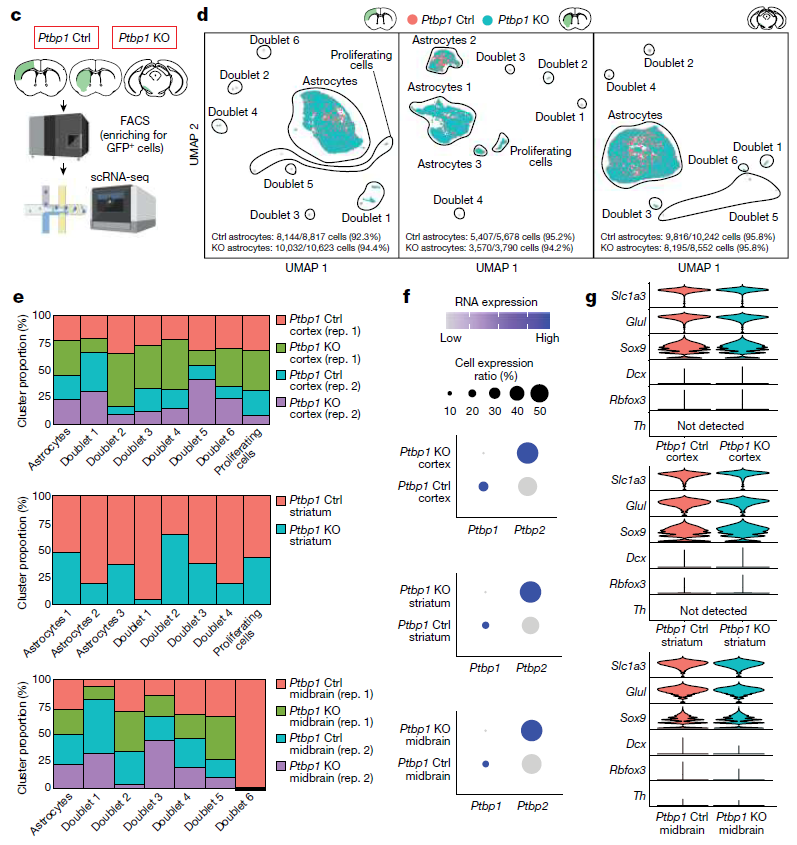
总的来说,该研究实现了对Ptbp1的高效和细胞特异性敲除,但无论是Ptbp1杂合缺失还是纯合缺失小鼠中,都没有观察到星型胶质细胞向神经元的转化。单细胞RNA测序(scRNA-seq)分析显示,杂合和纯合Ptbp1缺失的星形胶质细胞的基因表达有细微变化,但并未诱导神经元特异性基因表达或神经元样电生理特性。
结合最近发表的一系列论文均未能重复出敲低Ptbp1实现胶质细胞向神经元转化的结果,以及杨辉团队对自己此前 Cell 论文结果的否定,研究团队表示,此前的敲低Ptbp1后胶质细胞向神经元的转化的结果不太可能是Ptbp1功能缺失引起的。这也突出了在细胞类型转化的研究中使用遗传操作、谱系追踪和基因表达分析的重要性。
从论文的投稿记录来看,这篇论文于2021年9月就已投稿到 Nature,此后补充了大量实验和数据,从而更加严谨地论述了Ptbp1功能缺失不能诱导胶质细胞向神经元转化的观点。
而最早通过论文质疑Ptbp1作用的张春立教授直言不讳地表示:抑制Ptbp1就算有治疗潜力,那也不是源于胶质细胞向神经元的转化。
张春立教授在 Annual Review of Neuroscience 期刊发表了题为:Therapeutic Potential of PTBP1 Inhibition, If Any, Is Not Attributed to Glia-to-Neuron Conversion 的综述论文【7】。
张春立教授在这篇综述中回顾了基于Ptbp1的胶质细胞向神经元转化的研究争议,总结了对体内胶质细胞向神经元转化和Ptbp1未来研究的建议。

对于 Seth Blackshaw 团队在 Nature 发表的质疑文章,付向东团队同期在 Nature 发布了回应【8】。
付向东团队表示,在分析了 Seth Blackshaw 团队发布的单细胞RNA测序(scRNA-seq)数据后,得出了相反的结论。此外,之前在许多生物系统中的研究显示,敲除基因与敲低基因会产生不同的表型,因此,Ptbp1的敲除和敲低叶可能产生不同的表型。
虽然尚不能排除观察到的星形胶质细胞向神经元的转化可能是由于现有神经元泄漏造成的,但目前的scRNA-seq和免疫染色数据表明,存在一种星形胶质细胞或星形细胞样细胞亚类,其比成熟星形胶质细胞更倾向于重定向到神经元谱系,这一可能性有待于未来的研究。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
