
cuteSV——基因组结构变异检测工具
来源:NanoporeTechnologies 2020-11-15 09:59
基因组结构变异(Structural Variation,SV)包括缺失、插入、倒位、重复和易位等类型的基因组变异,与人类的疾病、进化、基因调控和相关表型等密切相关[1]。近年来,Oxford Nanopore Technologies等平台开发的长读长测序技术为准确检测基因组SV提供了可能。随着识别分辨率的不断提升,相比于短读长测序,长读长测序
基因组结构变异(Structural Variation,SV)包括缺失、插入、倒位、重复和易位等类型的基因组变异,与人类的疾病、进化、基因调控和相关表型等密切相关[1]。近年来,Oxford Nanopore Technologies等平台开发的长读长测序技术为准确检测基因组SV提供了可能。随着识别分辨率的不断提升,相比于短读长测序,长读长测序可检测出多达两倍的SV[2,3]。然而受限于SV检测的准确度、灵敏度及测序成本等因素(目前,大多数SV检测工具仍然高度依赖高深度的测序数据),在相关领域大范围推广SV检测技术仍然面临着巨大的挑战。
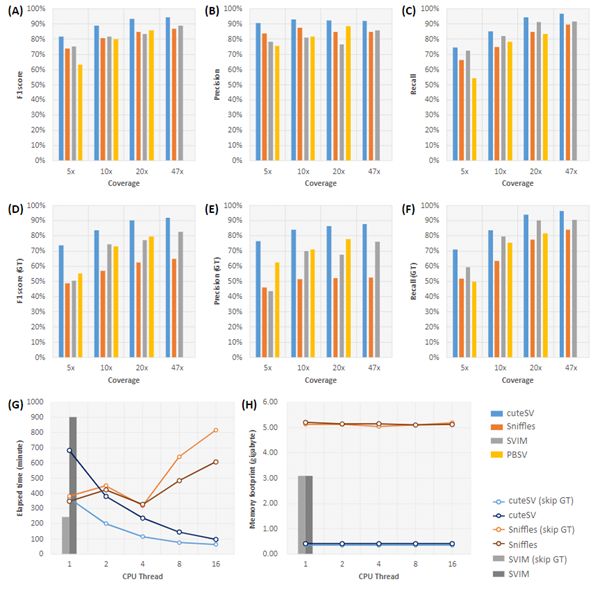
来自哈尔滨工业大学的姜涛老师在近期发表的文章中[4]介绍了一款全新的SV检测工具——cuteSV,该工具使用了纳米孔测序高通量测序平台PromethION最新发布的长读长测序技术,解析了Genome in a Bottle(GIAB)HG002人类参考基因组样本中的SV。cuteSV是一款快速的SV检测工具,在准确度和灵敏度等指标上均有优异的表现,同时在低测序深度数据上仍保持着良好的结构变异检测率(见图2)。这些结果表明,基因组SV研究领域正在进入一个高性能和低成本的时代。
cuteSV概览
cuteSV是一款基于长测序片段比对的基因组SV检测工具,它主要包括以下三个步骤(图1):
SV信号的识别:cuteSV设计和实现了多种SV信号识别方法,分别从比对内和比对间全面收集多种类型SV信号。同时,cuteSV采用了一套高效的启发式算法,将琐碎的插入与删除变异信号进行自动化整合以提升后续过程中SV识别的准确度。
SV信号的聚类:cuteSV设计了一种渐进复杂空间的聚类算法以及多重启发式的规则,在基因组局部区域内分别对SV信号进行聚类并对其进行优化,使零乱、繁杂的SV信号转换为高一致性的SV候选位点。
SV的检测与基因分型:cuteSV设计了一套特定的SV检测与基因分型规则,在上述聚类得到的SV候选位点中识别SV并完成基因分型。此外,cuteSV还可以输出基因型的似然估计、变异质量分数等多重统计量,以便于进一步的质量控制和构建更高准确度的检测集。
姜涛老师采用了最新发布的ONT PromethION HG002基因组样本数据集(平均读长:17335 bp,覆盖度:47x,下载地址:ftp://ftp.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/HG002_NA24385_son/UCSC_Ultralong_OxfordNanopore_Promethion/)对cuteSV的性能进行了基准测试[5],同时与Sniffles[6]、PBSV和SVIM[7]等工具进行了比较。cuteSV展现出了三大主要优势:
相较于其他工具,cuteSV在不同测序深度数据中能够检测出更多的SV(图2A-2C)。特别是在低覆盖度数据集中,cuteSV在准确度没有降低的前提下仍保持着较高的灵敏度。
cute SV在基因型检测方面表现出色,发现了更多的杂合与纯合SV。
与其他工具相比,cute SV具有更好的检测速度,同时内存的消耗也更低(图2H)。此外,cute SV的运行速度随CPU线程数的增加几乎呈线性增长,这表明其拥有良好的可扩展性。
在ONT PromethION数据的不同测序深度下,分别比较(A)F1分数、(B)准确度、(C)灵敏度、(D)结合基因型的F1分数、(E)结合基因型的准确度、(F)结合基因型的灵敏度。使用47x ONT PromethION数据分别在不同CPU线程下的(G)运行时间和(H)内存占用的统计结果。“GT”和“Skip GT”分别表示统计指标结合基因型和忽略基因型。由于SVIM不支持多线程运行,因此仅完成了单线程测试。PBSV在47x数据集的检测中程序异常终止,因此没有纳入统计。
纳米孔测序技术产生的超长读长序列,使大尺度结构变异的解析成为可能。图3给出了一个仅在纳米孔长读长序列中被检测出来的6481 bp的插入(断点位于chr1:9683994)。凭借纳米孔测序产生的超长读长的测序片段,片段比对工具在此区域CIGAR中给出了若干插入信号,cuteSV根据这些信号经过聚类检测出了该插入变异。然而其他长片段测序平台在此区域上产生的测序片段读长较短(平均长度7983 bp),片段比对工具无法比对出如此大的插入,因此导致了该插入变异的漏检。
展望
虽然长读长序列的可比对性远高于短读长序列,但由于测序错误、SV复杂程度、重复序列等因素的存在,片段比对仍存在着局限性。从头组装方法虽然克服了比对所产生的错误,且能够解析SV的单倍型,但从头组装方法仍然有着其自身的瓶颈,例如组装错误、计算量巨大、严重依赖不同类型的测序技术等。考虑到这些优缺点,姜涛老师认为基于比对和基于组装的检测方法是相辅相成的,将两种方法结合起来将有助于生成更高质量的SV检测集。
随着多元化大规模人群基因组项目的发展,群体基因组SV检测的需求也稳步增长。当前版本的cuteSV仅支持在单个基因组中检测SV,面向群体基因组进行SV检测对cuteSV来说仍是一个挑战。姜涛老师正在开发一个全新的群体基因组检测模块,旨在完成特定人群SV的检测。此外,姜涛老师所在的团队正在尝试建立一项新的基于长读长的变异检测技术,希望借此技术能够灵敏、高效地同时完成对SNP、Indel和SV的检测。(生物谷Bioon.com)
【直播预告】纳米孔测序在人类遗传学和罕见病研究中的应用
【日期】2020/11/19 15:00
http://count.medsci.cn/link/redirect/199d0462698595ba
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
