Science子刊:岳峰团队利用深度学习发现癌症中的新基因突变
来源:生物世界 2022-06-21 17:13
目前人们对结构变异的检测主要依赖于全基因组测序(WGS)。然而,由于 WGS 依赖于短测序,所以其在检测基因组重复区域结构变异上的能力十分有限。
肿瘤细胞的特征是基因组中有着大量的结构变异(Structural Variation,SV),比如大片段序列的缺失、扩增、倒位、易位等。很多结构变异已被证明和肿瘤的发生密切相关,并被用于肿瘤的分型与预后诊断,成为设计靶向治疗药物的重要依据,比如慢性髓系白血病中的 BCR/ABL1 融合基因。如何精准地在全基因组上检测结构变异一直是基因组学,病理学以及分子诊断的重点和难点。
目前人们对结构变异的检测主要依赖于全基因组测序(WGS)。然而,由于 WGS 依赖于短测序,所以其在检测基因组重复区域结构变异上的能力十分有限。而重复区域在人类基因组上的占比接近50%,所以 WGS 的结果有可能遗漏很多重要的结构变异。近年来发展起来的长序列测序方法,如 Nanopore 和 PacBio, 在一定程度上缓解了这一问题,然而这些方法通常需要很高的测序深度,而且输出结果经常达到几千甚至上万个结构变异,人们无法识别哪些变异更为重要。
来自美国西北大学的 Duane and Susan Burnham 讲席教授和癌症组学中心主任岳峰课题组及其合作者与2018年发表于 Nature Genetics 的工作首次开创性的利用全基因组染色质构象捕获技术(Hi-C)在不同肿瘤样本中对结构变异进行了全基因组识别【1】。Hi-C 最早被发明用来研究染色质的空间结构。由于结构变异能够在断点附近引发异常染色质交互,当把来自癌症细胞的 Hi-C 数据比对到参考基因组上时,不同种类的结构变异会在相应的基因组片段间形成独特的染色质交互模式。对这些不同交互模式的识别是利用 Hi-C 识别结构变异的基础。相比 WGS,Hi-C 对结构变异的识别不依赖于测序读段对结构变异断点的直接覆盖,因而能够在较低测序深度下检测位于复杂基因组区域的结构变异。然而,目前所有基于 Hi-C 的算法在识别结构变异的种类和大小上都有很大的局限性,包括目前使用最广泛的 Hi-C breakfinder【1】(由岳峰课题组共同开发),只能识别染色体间易位和超大片段(大于1Mb)的染色质内结构变异。这是因为,当只考虑染色质内交互,特别是小于1Mb染色质交互时,代表染色质固有结构的正常染色质互作会极大地干扰结构变异引发的交互信号,从而增加算法对小片段结构变异识别的难度。
2022年6月15日,岳峰课题组在 Science 子刊 Science Advances上发表了题为:EagleC: A deep-learning framework for detecting a full range of structural variations from bulk and single-cell contact maps 的研究论文【2】。
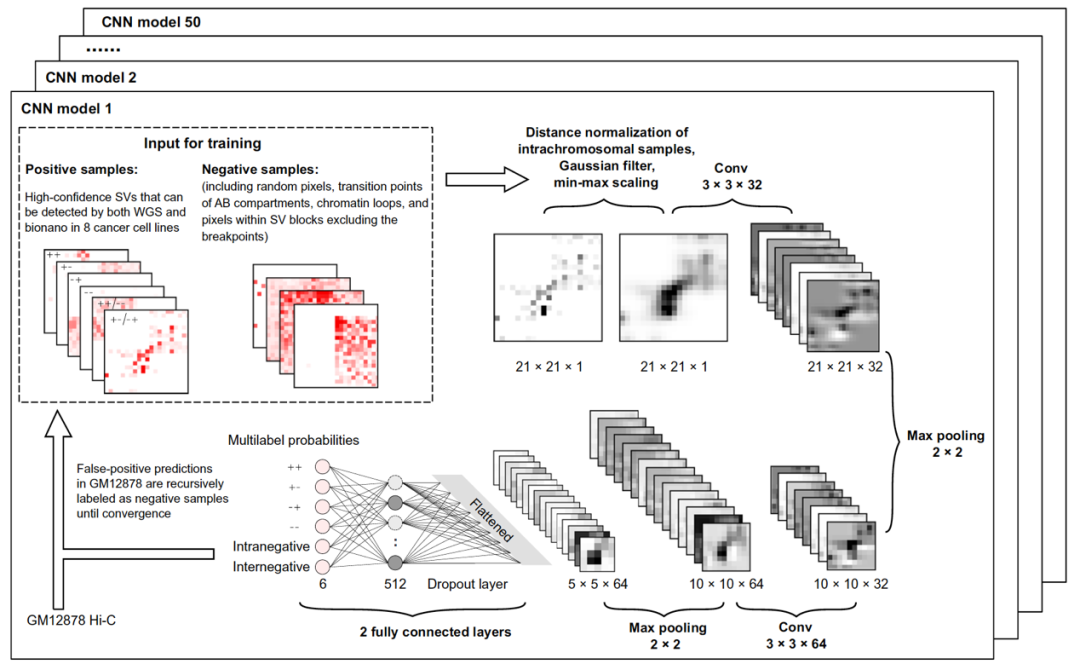
该研究基于深度学习和集成学习策略,提出一个全新的捕捉癌症基因组中结构变异的计算框架——EagleC。EagleC 能够用于多种染色质构象捕获技术,比如 Hi-C,Micro-C,HiChIP。更为重要的是,这个深度学习的模型可以直接用在单细胞 Hi-C 上,从而可以监测到癌症组织里的不同癌细胞结构变异的异质性。
EagleC 创造性地将深度学习(deep-learning)算法在图像识别上优势和集成学习(ensemble learning)相结合,以课题组先前在多个癌症细胞系中识别的高质量结构变异为训练样本,训练得到能够准确地在不同测序深度下识别结构变异的机器学习模型。为进一步降低预测的假阳性率,EagleC 采用了一种递归学习(iterative training)策略,在训练过程中不断从正常细胞 Hi-C 数据中引入阴性样本。相比已有基于 Hi-C 的算法,EagleC 能够检测出多一倍多结构变异数量,而且结构变异识别的准确率(precision)和召回率(recall)也都显著提高。另外由于 EagleC 能识别高分辨率的结构变异,该研究首次证实了 Hi-C 可以被用于在癌症样本中识别融合基因。
特别值得一提的是,由 EagleC 识别的结构变异大约有20-30%无法被目前常用的 WGS 和长片段测序如 nanopore 所识别。但是这些结构变异是可以被 RNA-Seq 数据所验证, 而且有相当一部分 Hi-C 检测到结构变异断点在已知癌症基因附近,所以很可能与这些基因的异常表达有关系。
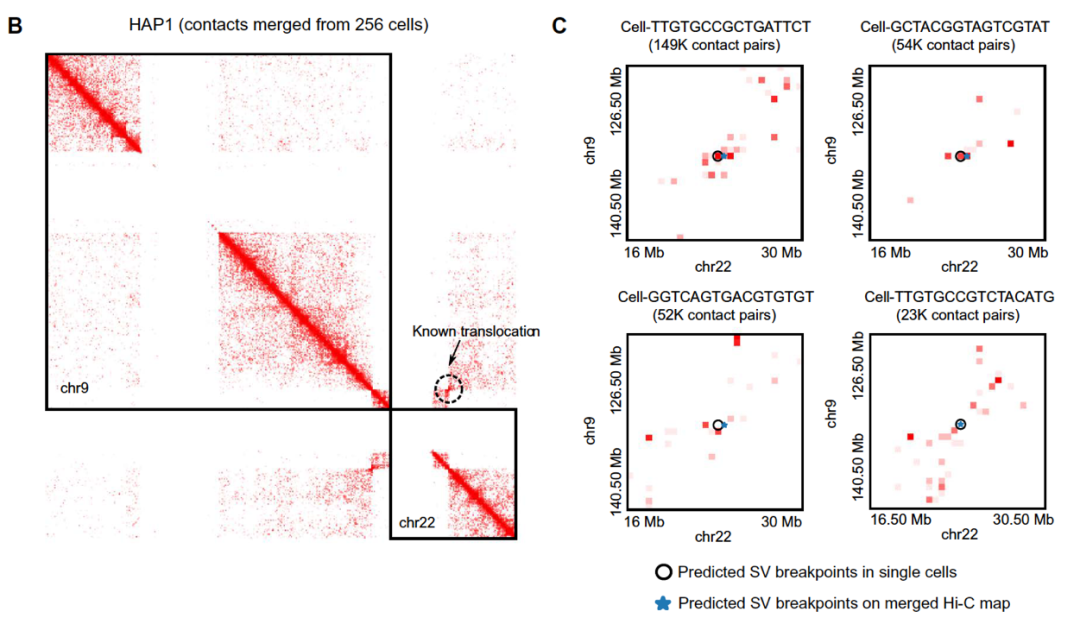
借助深度学习,EagleC 还能够利用其他染色质构象捕获技术,如 ChIA-PET、HiChIP、capture Hi-C 以及单细胞 Hi-C 等识别结构变异。更重要的是,EagleC 可以在癌症单细胞 Hi-C 数据上检测结构变异。如图下所示,EagleC 精确的在四个单细胞中检测到了慢性髓系白血病中的 BCR/ABL1 融合基因。所以 EagleC 将有望成为研究肿瘤病人样本的细胞异质性,理解肿瘤细胞发育和进化的一个重要计算方法。
最后,研究团队利用训练好的 EagleC 模型在超过100个癌症细胞系和病人样本中识别到了数千个结构变异。通过基因不研究发现,结构变异在肿瘤细胞中的形成与染色质的三维结构特征息息相关。在宏观尺度上,结构变异更容易发生在染色质区室A(Compartment A,代表活跃的染色质区域)之间;在局部尺度上,结构变异断点往往出现在染色质拓扑相关结构域(topologically associating domains,TAD)的边界附近。此外,研究还发现肿瘤相关基因的转录起始位点特异性地富集在位于 TAD 边界的结构变异断点附近。
综上所述,EagleC 是基于深度学习利用染色质构像捕获技术识别基因组结构变异的全新的计算框架,对发现新的基因融合事件,辅助肿瘤分型和靶向治疗药物的设计等具有潜在的应用价值。此外,EagleC 还能用于其他物种基因组的结构变异检测,进而评估参考基因组的组装质量。
版权声明
本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
87%用户都在用生物谷APP 随时阅读、评论、分享交流 请扫描二维码下载->