
研究揭示缩胆囊素受体识别配体和G蛋白选择性的分子机制
来源:上海药物研究所 2021-09-26 12:16
中国科学院上海药物研究所研究员蒋轶/徐华强团队、赵强团队、吴蓓丽团队、王明伟/杨德华团队和上海科技大学研究员赵素文团队在Nature Chemical Biology上,背靠背在线发表了题为Ligand recognition and G protein-coupling promiscuity of cholecystokinin
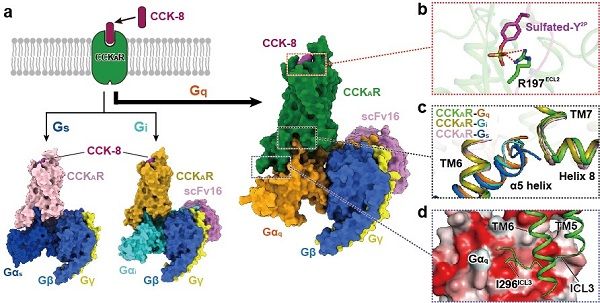
中国科学院上海药物研究所研究员蒋轶/徐华强团队、赵强团队、吴蓓丽团队、王明伟/杨德华团队和上海科技大学研究员赵素文团队在Nature Chemical Biology上,背靠背在线发表了题为Ligand recognition and G protein-coupling promiscuity of cholecystokinin A receptor和Structures of the human cholecystokinin receptors bound to agonists and antagonists的两项研究成果,解析了2种小分子拮抗剂(Devazepide、Lintitript)和激动剂NN9056与A型人源缩胆囊素受体(Cholecystokinin receptor,CCKAR)结合的3个晶体结构,以及多肽激素CCK-8的CCKAR分别与3种不同G蛋白(Gs、Gi、Gq)偶联复合物和多肽Gastrin-17分别与B型缩胆囊素受体(CCKBR)和两种G蛋白(Gi、Gq)形成复合物的5个冷冻电镜结构。两项研究揭示了多种多肽和小分子配体特异性识别缩胆囊素受体亚型的结构基础,阐明了配体选择性和受体活化的分子机制,破解了CCKAR选择性偶联不同G蛋白的奥秘,从而为靶向该类受体的药物研发提供了重要信息。
缩胆囊素(Cholecystokinin, CCK)是胃肠激素之一。CCK和胃泌素(Gastrin)是酪氨酰磺酸化多肽家族成员,具有保守的羧基末端八肽序列。两者在胃肠道和中枢神经系统中含量丰富,通过与缩胆囊素受体结合发挥激素调节和神经递质的作用。缩胆囊素受体包括A和B两个亚型(CCKAR和CCKBR),属于A类G蛋白偶联受体(GPCR)。CCKAR特异性识别磺酸化的CCK,而CCKBR对磺酸化和非磺酸化的CCK和Gastrin均有较强亲和力。这两类CCK受体参与调控的生理功能包括饱腹感、胰酶分泌和胆囊收缩,并与焦虑、记忆和药物成瘾等相关,因而是肥胖症、2型糖尿病和焦虑症等疾病的潜在治疗靶标。高选择性CCKAR非肽类拮抗剂(如Devazepide和Lintitript)被开发用来治疗胃肠功能紊乱、神经性疼痛和胰腺癌等疾病,而其激动剂NN9056对肥胖症具有一定疗效。但由于低药效和生物利用度等问题,大多数靶向CCKR的候选药物终止于临床研究。因此,开展CCKR的结构与功能研究将深化对其配体识别机制的认识,助力相关新药的创制。
GPCR被配体激活后主要通过偶联细胞内的G蛋白进行信号转导。根据α亚基的不同,G蛋白可分为Gi/o、Gs、Gq/11和G12/13d等四亚家族。虽然目前已报道了不同GPCR与其下游G蛋白结合的大量复合物结构,但受体如何精确识别这四种G蛋白尚不清楚。由于CCKAR能够偶联这四种不同的G蛋白,因而成为研究GPCR与G蛋白选择性结合机制的模式受体。
第一项研究中,科研人员解析了磺酸化CCK-8激活的CCKAR与Gq、Gs和Gi蛋白偶联的复合物冷冻电镜结构,分辨率分别为2.9 埃、3.1 埃和3.2 埃(图1a)。结合结构分析和功能验证,该工作展示了内源多肽激素—磺酸化CCK-8(以下简称CCK-8)的结合模式:CCK-8的氨基末端由受体的三个胞外环(ECL)紧紧包裹,其羧基末端插入受体的正构口袋。科研人员还鉴定了识别CCK-8的关键氨基酸残基和磺酸化基团发挥CCK-8活性的关键位点:位于CCKAR上ECL2区域的R197与CCK-8第二位酪氨酸修饰的磺酸基通过盐键结合进而决定磺酸化多肽的高亲和力(图1b)。
此外,该研究也对CCKAR选择性偶联Gq、Gs和Gi的分子机理进行了阐述,并提出ICL3参与CCKAR与Gq偶联的新机制。科研人员发现,偶联三种不同G蛋白的受体呈现相似的激活构象,然而受体与不同G蛋白结合界面的面积显示Gq > Gs > Gi的趋势。与之一致,CCKAR与其主要下游信号蛋白Gq偶联时产生的最大的激活效应(Emax)显着高于Gs和Gi。上述结果支持Gq是CCKAR主要偶联的G蛋白类型,明确了接触面积在G蛋白选择性识别中的作用。该工作进一步证实了Gq、Gs和Gi蛋白α亚基α5螺旋的末端弯钩(Wavy hook)是CCKAR对G蛋白选择性的参与者(图1c)。在CCKAR与Gq的复合物中,科研人员还发现CCKAR胞内环ICL3的I296与Gαq亚基的一个疏水结合口袋结合,这是首次在GPCR结构中观测到ICL3与Gα亚基的相互作用方式(图1d)。相反,由于Gαs和Gαi与Gαq在该疏水口袋残基组成的差异,CCKAR的相应ICL3区域与Gs和Gi不存在该疏水作用,说明ICL3决定了G蛋白之选择性。
第二项研究中,科研人员解析了CCKAR与小分子拮抗剂Devazepide、Lintitript和激动剂NN9056结合的三个晶体结构,以及结合多肽Gastrin的CCKBR分别与Gi和Gqo偶联的两个复合物冷冻电镜结构(图2a-e)。该工作揭示出多肽和小分子拮抗剂识别CCKR的分子机制,发现ECL2是CCKAR和CCKBR选择性识别多肽配体的决定因素。ECL2上的H207与多肽中D2形成的氢键决定了CCKBR对CCK-8、CCK-8ns和Gastrin-17的高亲和力(图2f),而ECL2上的R197与多肽中Y7的磺酸基形成的盐键决定了CCK-8对CCCKAR的高选择性(图2g)。同时,科研人员还发现N3336.55和R3366.58在CCKAR识别Devazepide和Lintitript过程中发挥了关键作用,为后继选择性靶向药物的开发奠定了基础。
此外,科研人员通过比对CCKAR结合拮抗剂Devazepide的结构、CCKAR结合激动剂NN9056的结构以及CCKAR同时结合激动剂CCK-8ns和Gq蛋白的结构,结合分子动力学模拟实验,阐释了CCKAR逐步激活的过程。相对于拮抗剂,激动剂在结合口袋中结合更深(图2h),随后引起PIF和FxxCWxP保守基序的变化(图2i和2j),进而导致受体胞内部分发生TM6和TM5向外以及TM7向内移动的激活态构象变化,从而揭示了CCKAR逐步激活的分子机制。(生物谷Bioon.com)
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
