
Science:利用单细胞染色质可及性图谱揭示原发性人类癌症中的恶性调控程序
来源:生物谷原创 2024-09-27 13:48
多种人类原发性癌症类型的 TCGA 单细胞图谱和用于解释癌症中顺式调控元件的可解释深度学习模型,为了解导致原发性人类癌症恶性表型的分子程序提供了新的资源。
基因病变是癌症的根本原因,也是基因调控变化的驱动力,而基因调控变化是癌症的经典表型“特征”。癌症基因组图谱(TCGA)旨在了解与这些基因突变相关的多种分子特征,以及驱动不同癌症类型表型的相关基因调控变化。
一种识别与基因表达相关的调控元件和反式因子的方法是利用转座酶可接触染色质测序(ATAC-seq)绘制染色质可及性图谱,其中ATAC-seq可识别基因组中被转录因子(TF)和聚合酶等调控蛋白主动结合的部分。虽然 TCGA 以前曾在“大块”癌症组织中研究过这些调控元件,但这种分析将癌症的恶性特征与相关基质细胞或免疫细胞中存在的调控元件景观联系在一起。相比之下,单细胞染色质可及性数据可对这些信号进行解卷积。
此外,通过使用深度学习模型,可以从这些癌症特异性数据中学习到癌症特异性基因调控“语法”,从而识别与转录因子结合相关的基因组序列,并预测癌症相关突变对这些调控元件可及性的影响。

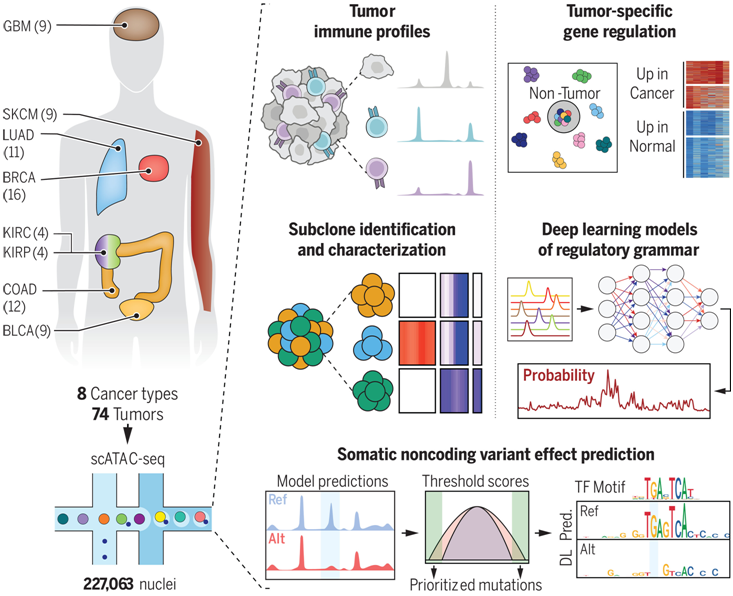
在一项新的研究中,来自斯坦福大学等研究机构的研究人员利用 TCGA 存档样本生成了单细胞染色质可及性图谱,该图谱涵盖 8 种癌症类型和 74 例癌症样本(由 227063 个细胞核组成)。通过标记基因周围的基因可及性可以让他们识别癌细胞与肿瘤浸润基质细胞和免疫细胞。与拷贝数改变相关的染色质可及性信号的兆碱基尺度变化根据不同的拷贝数变化确定了肿瘤亚克隆。他们观察到肿瘤亚克隆会表现出与受亚克隆特异性拷贝数改变影响的转录因子相关的不同基因调控程序。相关研究结果发表在2024年9月6日的Science期刊上,论文标题为“Single-cell chromatin accessibility reveals malignant regulatory programs in primary human cancers”。
利用这些数据,他们训练了可解释的基于神经网络的顺式调控模型,该模型指定了与癌症(与正常组织相比)和癌症亚型中不同染色质可及性信号相关的特定转录因子基序。这项分析提供了一个平台,可将癌细胞的调控语法和调控模块与最相似的健康组织类型的调控语法和调控模块进行比较。对乳腺癌亚型的分析表明,基底样乳腺癌亚型的染色质特征与分泌型管腔上皮细胞最相似,从而证实了这种分子亚型与基底型正常细胞在表观遗传学上并不最相似。他们利用他们的神经网络模型,通过将在人类癌症或人群中观察到的单核苷酸变异和癌症特异性体细胞突变匹配到基因组中,就可预测遗传变异对染色质可及性的影响。与匹配的非癌症相关基因相比,预测会导致染色质可及性强烈增益或缺失的突变富集在癌症相关基因附近,这提供了体细胞非编码突变可驱动癌症相关染色质调控变化的证据。
图片来自Science, 2024, doi:10.1126/science.adk9217
综上所述,多种人类原发性癌症类型的 TCGA 单细胞图谱和用于解释癌症中顺式调控元件的可解释深度学习模型,为了解导致原发性人类癌症恶性表型的分子程序提供了新的资源。(生物谷Bioon.com)
参考资料:
Laksshman Sundaram et al. Single-cell chromatin accessibility reveals malignant regulatory programs in primary human cancers. Science, 2024, doi:10.1126/science.adk9217.
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
