
内质网应激通过miR-188-5p/hnRNPA2B1介导的肝细胞癌PKM2上调促进索拉非尼耐药
来源:本站原创 2022-01-19 14:32
越来越多的证据表明,内质网(ER)应激促进了肝细胞癌(HCC)对索拉非尼的耐药性。然而,人们对其潜在机制知之甚少。本研究的目的是探讨内质网应激促进肝癌细胞对索拉非尼耐药的机制。
越来越多的证据表明,内质网(ER)应激促进了肝细胞癌(HCC)对索拉非尼的耐药性。然而,人们对其潜在机制知之甚少。本研究的目的是探讨内质网应激促进肝癌细胞对索拉非尼耐药的机制。

图片来源: https://doi.org/10.1016/j.omtn.2021.09.014.
能量代谢重新编程也被报道可以提高肿瘤细胞的存活率和化疗耐药性。能量代谢重编程,特别是由PKM2持续激活介导的异常激活的糖酵解在肿瘤细胞对索拉非尼耐药中的作用尚不清楚。研究表明,PKM2在肺癌、乳腺癌、结直肠癌等多种肿瘤中均有过表达,并与细胞增殖、凋亡抵抗、侵袭和迁移密切相关。
作者的研究表明,PKM2在肝细胞癌中显著过表达,而PKM基因在正常肝细胞中的表达产物PKM1在肝癌中几乎不表达。PKM2在细胞凋亡抵抗和化疗耐药中起重要作用。此外,作者的研究还发现,肝癌组织中PKM2的过表达对索拉非尼耐药有一定的贡献。提示肝癌组织中PKM2的过度表达和充分激活可加速糖酵解过程,迅速有效地为肝癌细胞提供充足的ATP,使部分肝癌细胞逃脱索拉非尼诱导的细胞凋亡。
据报道,内质网应激通过减少细胞表面葡萄糖转运蛋白1(GLUT1)的表达来调节细胞内葡萄糖和乳酸代谢,从而增加正常细胞的死亡。内质网应激是否影响糖代谢重编程,尤其是PKM基因的表达,是我们的主要研究方向。
作者首先在肝癌组织标本中发现PKM2与ER应激密切相关;此外,作者还发现ER应激刺激物TM增加了肝癌细胞中PKM2的表达,而用siRNA沉默PKM2则降低了ER应激标记蛋白GRP78、PERK、IRE1和ATF6的表达。
内质网应激可上调PKM2的表达,提示内质网应激作为细胞糖代谢重编程的诱导因子,可充分激活糖酵解,使肿瘤细胞直接有效地获得ATP,并为应激细胞提供降解错误折叠和未折叠多肽片段所需的补偿性能量。
据报道,内质网应激通过诱导或下调miRNAs的表达来影响细胞的生物学行为。差异表达的miRNAs通过沉默相关靶基因的表达间接调节肿瘤细胞的增殖、分化和凋亡。结直肠癌中上调的miR-27A与糖酵解密切相关,通过抑制AMPK和增强哺乳动物雷帕霉素靶点(MTOR)信号转导化疗耐药。
关于miRNAs是否参与内质网应激调节PKM2表达的研究有限。在本研究中,作者观察到17个miRNAs在内质网应激下的肝癌细胞中差异表达,其中4个与肿瘤细胞的能量代谢重编程有关-miR-188-5p、miR33b-3p、miR-22-5p和miR-301a-5p。此外,作者的结果显示miR-188-5p在内质网应激的肝癌细胞中的下调最为显著。
生物信息学分析表明,PKM2不是miR-188-5p的直接靶基因。MiR-188-5p是否通过靶向PKM2的直接靶基因来调节PKM2的激活是我们的研究方向。在miR-188-5p的预测靶基因中,作者发现hnRNP家族,包括hnRNPA0、hnRNPA1和hnRNPA2B1,可能通过剪接RNA来调节PKM基因的选择性表达。
在本研究中,作者通过双荧光素酶报告基因和生物信息学分析,进一步确定nRNPA2B1是miR-188-5P的直接靶基因。HnRNPA2B1是一种RNA结合蛋白,参与多种生物学过程,如将异质核RNA加工成成熟的mRNA、RNA剪接、基因表达的反式激活和蛋白质翻译的调节。
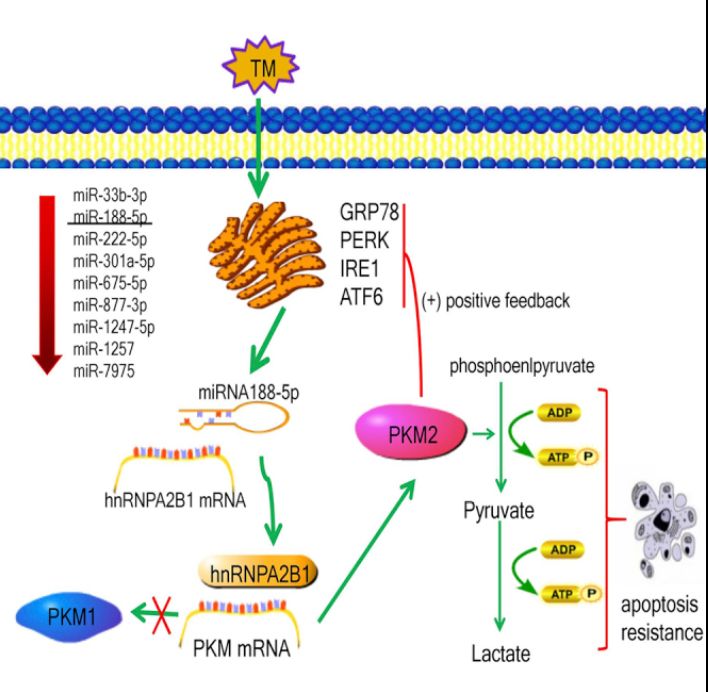
靶向miR-1885p/hnRNPA2B1/PKM2通路和内质网应激可能有助于克服肝癌治疗中索拉非尼的耐药性
图片来源: https://doi.org/10.1016/j.omtn.2021.09.014.
综上所述,本研究证实了肝癌组织中激活的内质网应激与过表达的PKM2之间的相关性,两者均与索拉非尼耐药有关,并揭示了内质网应激可通过miR-188-5p/hnRNPA2B1上调PKM2的表达。
内质网应激肝癌细胞中抑癌基因miR-1885p的下调可通过直接靶向hnRNPA2B1增加PKM2介导的索拉非尼耐药性。因此,靶向miR-188-5p/hnRNPA2B1/PKM2通路和内质网应激可能有助于克服肝癌治疗中索拉非尼的耐药性。(生物谷 Bioon.com)
参考文献
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
