
2025年,这20款重磅新药有望在中国获批
来源:医药魔方 2024-12-28 11:22
2024年即将结束,截至目前,国家药品监督管理局今年共批准了74款新药。
2024年即将结束,截至目前,国家药品监督管理局今年共批准了74款新药(不包括中药、新适应症),其中包含多款FIC创新药,在肿瘤领域,包括全球首个针对叶酸受体α阳性卵巢癌的ADC药物索米妥昔单抗、PD-1/VEGF双特异性抗体依沃西单抗以及Nectin-4 ADC维恩妥尤单抗等;自免领域有IL-17A/F单抗比吉利珠单抗获批用于强直性脊柱炎;另外罕见病领域有诺华研发的补体因子B抑制剂获批用于阵发性睡眠性血红蛋白尿症(PNH)等,这些药品的上市将为中国患者带来新的治疗机会。
在2025即将到来之际,医药魔方为大家总结梳理一下明年有望在中国上市的重磅药品,供大家参考。
2025年有望在中国获批的重磅新药
一、肿瘤领域
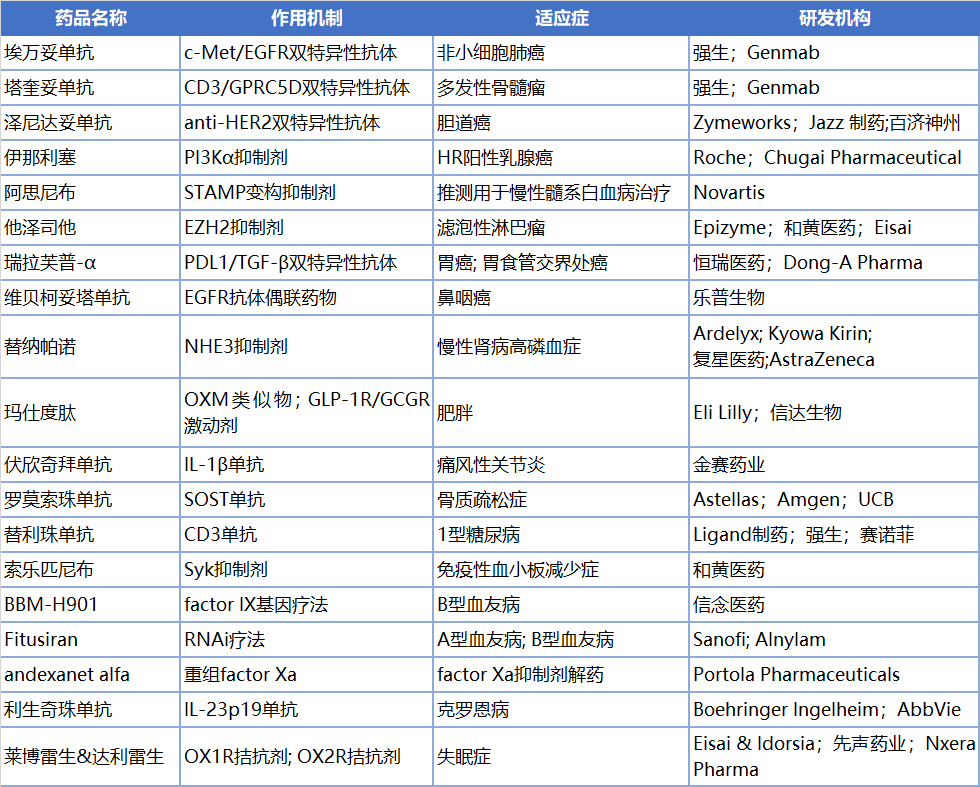
埃万妥单抗(amivantamab,Rybrevant)
预计批准时间:2025Q1
埃万妥单抗是由强生和Genmab开发的一款first-in-class的c-Met/EGFR双特异性抗体。最早于2021年5月经FDA加速批准用于治疗正在接受或接受过含铂化疗后仍有疾病进展的EGFR Exon 20突变的非小细胞肺癌成年患者。今年3月FDA批准其联合化疗用于EGFR Exon 20突变的NSCLC患者的一线治疗,这是FDA批准用于一线治疗此类患者的首款疗法,并获得NCCN指南一类推荐。

数据来源:NextPharma®数据库-新闻热点
中国进度方面,强生于2023年10月首次递交埃万妥单抗的上市申请;今年1月向CDE同时递交兰泽替尼和埃万妥单抗的上市申请,推测申报适应症为二者联合治疗EGFR突变局部晚期或转移性NSCLC患者。III期临床MARIPOSA研究结果显示,对于EGFR突变患者,埃万妥单抗联合兰泽替尼与疗法相较于奥希替尼,可将疾病死亡风险降低30%,并将患者的中位无进展生存期(mPFS)延长7个月。此外,强生于2024年9月向CDE递交埃万妥单抗皮下注射剂的上市申请,目前处于排队待审状态。预计2025Q1埃万妥单抗有望在中国获批。
塔奎妥单抗(talquetamab,Talvey)
预计批准时间:2025Q2
塔奎妥单抗是一款CD3/GPRC5D双特异性抗体, 也是首个针对GPRC5D靶点获批的药物。GPRC5D属于G蛋白偶联受体,多表达于多发性骨髓瘤,在正常组织的表达仅限于皮肤(毛囊和小汗腺)和睾丸(输精小管),且其表达水平与靶点BCMA相对独立。
2023年8月,FDA基于II期MonumenTAL-1研究结果加速批准塔奎妥单抗用于治疗患有复发性或难治性多发性骨髓瘤的成年患者,这些患者之前至少接受过4种疗法,包括蛋白酶体抑制剂、免疫调节剂和抗 CD38 单克隆抗体,结果显示,每两周皮下注射给药0.8 mg/kg时,ORR达到73.6%。
2024年欧洲血液学会EHA会议上,首次披露了塔奎妥单抗在中国人群中的有效性数据,结果显示,0.4 mg/kg QW、0.8 mg/kg Q2W队列的ORR分别是69.0% 和66.7%,VGPR率均大于58%,首次缓解的中位时间为1.3个月;安全性方面,与味觉、皮肤和指甲相关的 AE 主要为 1/2 级,且味觉相关的不良事件率为(25.0–41.1%),低于全球(71.0–72.0%)。

数据来源:NextPharma®数据库-临床结果
2024年2月,塔奎妥单抗在中国首次申请上市并纳入优先审评,用于单药治疗既往接受过至少三种治疗(包括一种蛋白酶体抑制剂、一种免疫调节剂和一种抗CD38抗体)的复发或难治性多发性骨髓瘤(R/R MM)成人患者,预计2025年Q2有望在中国上市。
泽尼达妥单抗(zanidatamab,Ziihera)
预计批准时间:2025Q2
泽尼达妥单抗是一种基于 Zymeworks 专有 Azymetric™平台研发的 HER2 靶向双特异性抗体,能够同时结合 HER2 受体的两个非重叠表位,这种独特设计可以引起双重HER2信号阻断。2018年,Zymeworks与百济神州达成交易,授予百济神州在亚洲(除日本外)、澳大利亚和新西兰的开发和商业化权益;2022年10月,Jazz制药宣布获得其他国家地区开发和商业化泽尼达妥单抗的独家许可。

数据来源:NextPharma®数据库-医药交易
Jazz制药于今年11月宣布泽达尼妥单抗获FDA加速批准,用于治疗既往经治的HER2阳性(IHC3+)胆道癌(BTC),该项获批基于HERIZON-BTC-01试验数据,在既往接受过治疗的不可切除、局部晚期或转移性的HER2阳性(IHC3+)BTC患者中,主要终点ORR达52%,次要终点DOR为14.9个月。安全性方面,53%患者会发生严重不良反应,20%以上的患者最常见的不良反应是腹泻、输液相关反应、腹痛和疲劳。
今年6月,基于HERIZON-BTC-01试验结果,百济神州递交泽尼达妥单抗在中国的首个上市申请,用于治疗既往接受过全身治疗的HER2高表达的不可切除局部晚期或转移性胆道癌患者,该项适应症曾于2023年11月被CDE纳入优先审评,预计2025Q2有望获批。
伊那利塞(inavolisib,Itovebi)
预计批准时间:2025Q3
伊那利塞是罗氏开发的一款口服高选择性PI3Kα抑制剂,针对PIK3CA突变、HR阳性乳腺癌。PIK3CA突变在乳腺癌中非常普遍,发生在约40%的HR阳性肿瘤中,这款产品的突出优势在于高选择性,它对PI3Kα的选择性超过其他亚型(PI3K β/δ/γ)300倍,超过PIK家族成员2000倍。目前伊那利塞在中国同赛道排名第一,全球同赛道排名中位于第二,仅次于诺华的阿吡利塞。
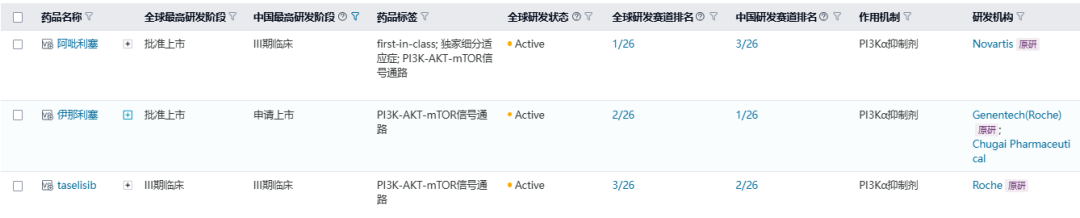
数据来源:NextPharma®数据库-赛道排名
今年5月,FDA授予伊那利塞突破性疗法认定,与哌柏西利和氟维司群联合用于一线治疗PIK3CA突变的HR+/HER2-局部晚期或转移性乳腺癌成人患者。BTD认定基于一项国际多中心3期INAVO120(NCT04191499)研究,临床结果显示在一线治疗中,与单独使用派柏西利和氟维司群相比,联合伊那利塞治疗可将疾病恶化或死亡风险降低 57%,中位PFS得以延长(15.0个月 vs. 7.3个月;HR=0.43, p<0.0001)。10月10日,伊那利塞在美国获常规批准上市。
目前,伊那利塞的另外两项3期临床研究正在招募患者,用于评估伊那利塞在不同组合疗法中的获益情况,分别为:1)伊那利塞+氟维司群对比阿吡利塞+氟维司群用于二线治疗PIK3CA突变、HR+/HER2-局部晚期或转移性乳腺癌患者的安全性和有效性研究(INAVO121);2) 伊那利塞联合Phesgo皮下注射剂(曲妥珠单抗+帕妥珠单抗+重组人玻璃酸酶)对比安慰剂联合Phesgo,用于先前未经治疗的HER2+晚期乳腺癌患者维持疗法的安全性和有效性研究(INAVO122)。
目前伊那利塞是中国研发进度最快的选择性PI3Kα抑制剂,于今年6月向CDE提交上市申请,并被纳入优先审评目录,有望2025Q3在中国获批。
阿思尼布(asciminib,Scemblix)
预计批准时间:2025Q3
阿思尼布是由诺华开发的一款first-in-class的STAMP(Bcr-Abl myristoyl pocket)变构抑制剂,不同于结合催化位点的ABL1激酶抑制剂,阿思尼布能够特异性结合ABL豆蔻酰基口袋,诱导ABL1形成非活性激酶构象从而发挥作用。
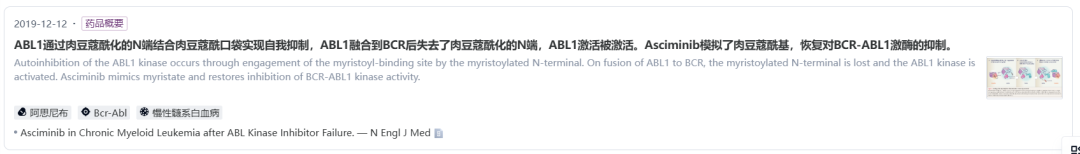
数据来源:NextPharma®数据库-知识卡片
2021年2月,阿思尼布获得FDA突破性疗法认定,同年10月,FDA加速批准阿思尼布用于既往接受过两种或多种酪氨酸激酶抑制剂治疗的费城染色体阳性慢性粒细胞白血病(Ph+ CML-CP)成年患者;并完全批准其用于具有T315I突变的Ph+ CML-CP成人患者。由于阿思尼布与Bcr-ABL1结合的位点不同于现有TKI(如伊马替尼、尼洛替尼),阿思尼布显示出与结合于催化位点TKI不同的耐药谱,为一些CML患者(如ABL1 T315I突变患者)带来全新的治疗选择。2024年10月FDA又加速批准阿思尼布用于新诊断的Ph+ CML-CP成年患者。
阿思尼布的全球多中心注册性3期临床研究(NCT04971226)结果显示,对于新诊断的CML患者,阿思尼布单药治疗的MMR达到67.7%,而接受其他TKI(伊马替尼或第二代TKI)治疗的患者MMR仅为49.0%; 同时阿思尼布组的患者发生3 级以上不良事件的概率也小于伊马替尼和第二代TKI组的患者。2024年6月诺华公司向NMPA递交了阿思尼布用于治疗CML的上市申请,预计2025Q3获批。
他泽司他(tazemetostat,Tazverik)
预计批准时间:2025Q3
他泽司他是Epizyme开发的一款first-in-class的EZH2甲基转移酶抑制剂,最早于2020年1月经FDA加速批准用于治疗16岁及以上患者转移或局部晚期上皮样肉瘤患者,是FDA批准的唯一一款EZH2抑制剂,也是第一个获批用于上皮样肉瘤的药物。

数据来源:NextPharma®数据库-疾病相关药品
2020年6月FDA加速批准他泽司他用于既往至少接受过二线治疗或无其他疗法的EZH2+复发/难治性滤泡性淋巴瘤患者。此批准基于2期试验(NCT01897571)的结果,他泽司他在EZH2+ 复发/难治性滤泡性淋巴瘤患者中的ORR达到69%。
2021年8月和黄医药以总金额3.75亿美元从Epizyme获得了在大中华区开发及商业化他折司他的权益。2022年和黄医药完成了他泽司他联合来那度胺以及利妥昔单抗治疗复发/难治性滤泡性淋巴瘤患者的中国桥接研究(NCT04224493)的患者入组,同年5月,其临床急需进口药品申请在海南批准,用于治疗某些上皮样肉瘤和滤泡性淋巴瘤患者。
今年7月和黄医药向CDE递交了他泽司他用于治疗复发或难治性滤泡性淋巴瘤成人患者的上市申请,并获得了优先审评资格,预计2025Q3有望获批。
瑞拉芙普-α(SHR-1701)
预计批准时间:2025Q4
瑞拉芙普-α是恒瑞医药研发的一款PD-L1/TGF-βRII双功能融合蛋白,由靶向PD-L1的IgG4单抗融合TGF-βIIR胞外结构域而成,在抑制PD-1/PD-L1 通路的基础上,靶向中和肿瘤微环境中的TGF-β可以进一步使 T细胞恢复活性,增强免疫应答。
2024年9月,恒瑞宣布瑞拉芙普-α上市申请获国家药监局受理,联合氟尿嘧啶类和铂类药物用于局部晚期不可切除、复发或转移性胃及胃食管结合部腺癌的一线治疗。目前以氟尿嘧啶类和铂类为基础的联合化疗是我国晚期胃癌一线治疗方案之一,但化疗给患者带来的长期生存获益有限。免疫检查点抑制剂单药治疗胃癌时,疗效欠佳。目前,免疫检查点抑制剂联合化疗成为晚期胃癌患者的一线治疗新标准。
今年ESMO大会上,恒瑞公布了一项SHR-1701联合化疗用于晚期一线HER2阴性胃或胃食管结合部腺癌(G/GEJA)的3期临床数据(NCT04950322),结果显示,SHR-1701联合化疗一线治疗HER2阴性G/GEJA患者总生存期获益显著,在PD-L1 CPS≥5人群中,SHR-1701组和安慰剂组中位生存期分别为16.8个月和10.4个月;ITT人群中,SHR-1701组和安慰剂组相比,同样达到主要终点。
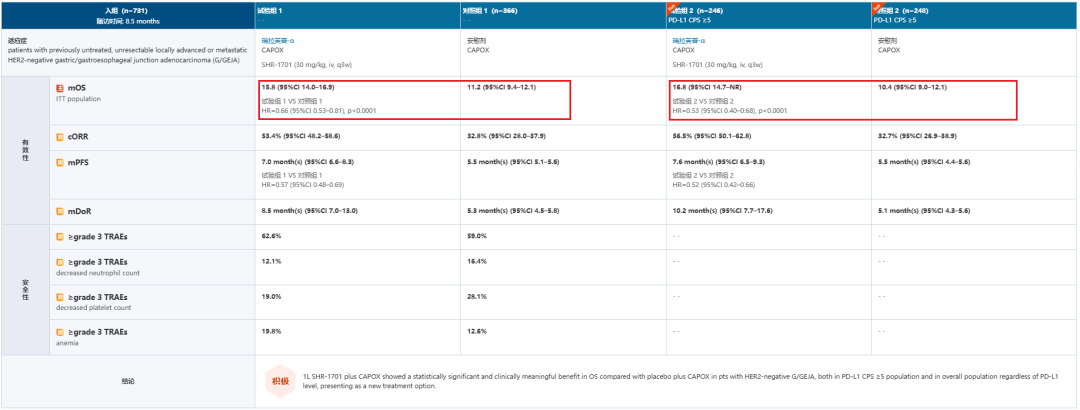
数据来源:NextPharma®数据库-临床结果
目前瑞拉芙普-α是全球进展最快的PD-L1/TGF-βRII双功能融合蛋白,国内外尚无同类产品获批上市,预计2025Q4在中国获批。
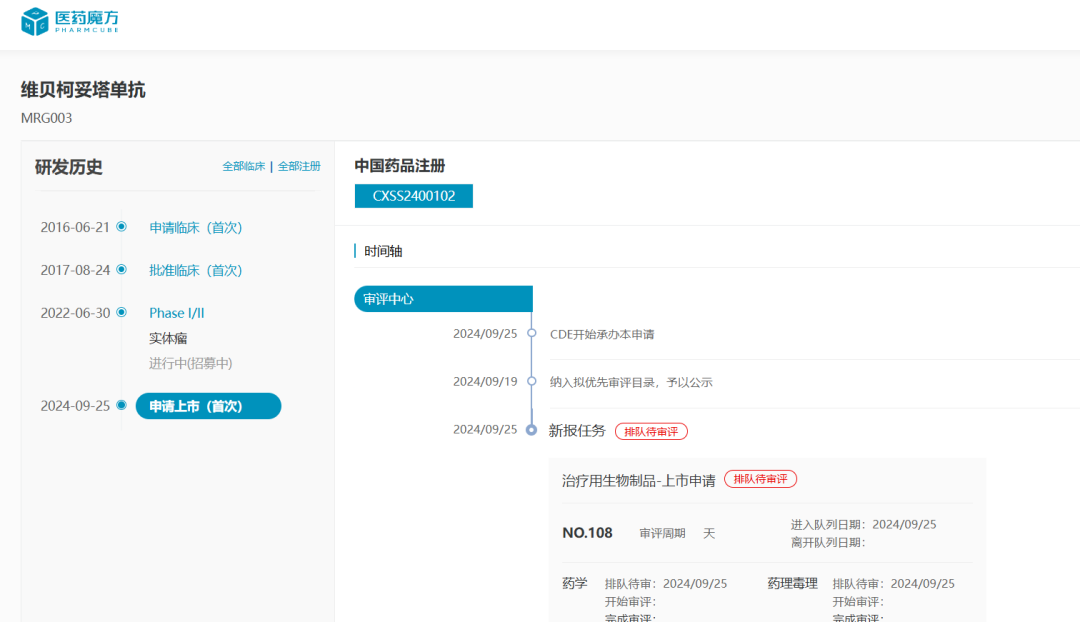
维贝柯妥塔单抗(MRG003)
预计批准时间:2025Q4
维贝柯妥塔单抗是由乐普生物研发的靶向EGFR抗体偶联药物,由人源化EGFR IgG1单抗、可降解缬氨酸-瓜氨酸linker、以及微管抑制剂MMAE组成,能以高亲和力特异性结合肿瘤细胞表面的EGFR,内吞后通过蛋白酶裂解释放payload,从而导致肿瘤细胞死亡。根据NextPharma®数据库统计,目前全球范围内共有40款靶向EGFR的活跃ADC管线,维贝柯妥塔单抗是该赛道中国进度最快的产品,有望率先抢占鼻咽癌市场。
2024年12月7日,乐普生物在ESMO Asia大会上公布了MRG003联合PD-1抗体普特利单抗治疗EGFR突变、复发或转移性鼻咽癌的2期临床(NCT05688605)数据,结果显示MRG003联合普特利单抗cORR达到66.7%,安全性与有效性良好,有望为抗PD-(L)1治疗失败的复发或转移性鼻咽癌患者带来治疗选择。
自2022年9月开始,MRG003已分别获得CDE突破性疗法认定、FDA孤儿药资格认证、FDA快速通道资格及BTD认定。今年9月,维贝柯妥塔单抗首个NDA被CDE受理,作为PD-1/PD-L1抑制剂治疗失败的复发/转移性鼻咽癌患者三线疗法,目前处于排队待审状态,预计2025Q4在中国上市。
数据来源:NextPharma®数据库-中国研发进度
二、内分泌及代谢领域
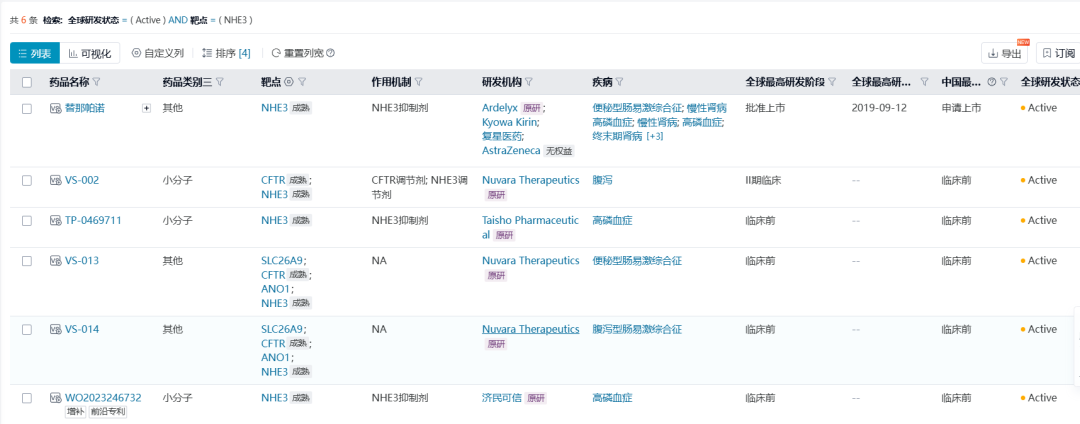
替那帕诺(tenapanor,Xphozah)
预计批准时间:2025Q1
替那帕诺是由Ardelyx原研的唯一一款在全球获批上市的NHE3小分子抑制剂。替那帕诺是一种磷吸收抑制剂,通过阻断NHE3增加细胞内质子,从而减少肠道的磷吸收,同时增加粪便中钠离子和水的含量,因此替那帕诺会造成患者腹泻。
数据来源:NextPharma®数据库-基础检索
2021年替那帕诺用于慢性肾病(CKD)成年患者血磷控制的上市申请被FDA拒绝。FDA指出虽然替那帕诺能够有效降低CKD患者的血磷,但是幅度很小,临床意义尚不明确。在Ardelyx重新提交NDA后,2023年FDA批准了替那帕诺作为附加治疗用于降低成年CKD患者的血磷水平。此外,FDA还批准了替那帕诺用于那些对磷结合剂反应不佳或不耐受的成年CKD患者,以帮助控制其血磷水平。
替那帕诺在中国的进展也值得关注。2017年复星医药以总金额1.25亿美元从Ardelyx手中获得了替那帕诺的在中国内地、香港及澳门特别行政区的独家临床开发和商业化权益。2023年7月复星医药向NMPA递交替那帕诺用于治疗慢性肾病血透患者高磷血症的上市申请。替那帕诺在中国的III期注册性临床(CTR20202588)的结果显示,替那帕诺治疗组中44.59%的患者的血磷水平降到了5.5 mg/dL以下。预计2025Q1有望在中国获批。
玛仕度肽(mazdutide,IBI362)
预计批准时间:2025Q2
玛仕度肽是信达生物与礼来开发的一款胰高血糖素样肽-1受体(GLP-1R)/胰高血糖素受体(GCGR)双重激动剂。作为一种哺乳动物胃泌酸调节素(OXM)类似物,玛仕度肽除了可通过激动GLP-1R促进胰岛素分泌、降低血糖和减轻体重外,还可通过激动GCGR增加能量消耗而增强减重疗效,同时改善肝脏脂肪代谢。
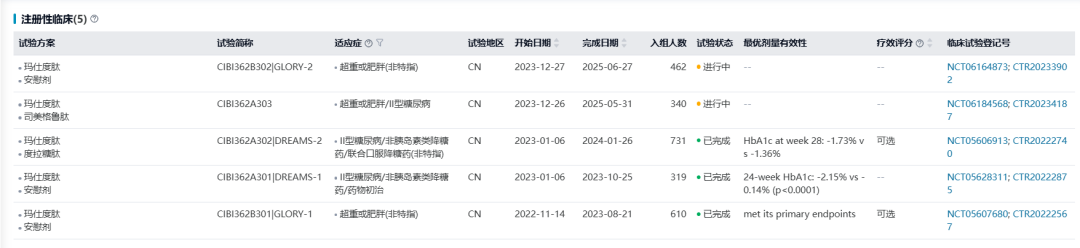
玛仕度肽在中国超重或肥胖成人受试者中的首个III期临床GLORY-1研究达成主要终点和所有关键次要终点。玛仕度肽6mg组受试者治疗32周后体重相对基线的百分比变化显著优于安慰剂组(-13.38% vs -0.24%)。
2024年7月,信达生物宣布玛仕度肽在中国2型糖尿病受试者中开展的III期临床研究(DREAMS-1)达到主要终点和全部关键次要终点,展现出降糖、减重双达标及心血管肾脏代谢指标的综合获益,第24周时,玛仕度肽4mg组和6mg组HbA1c较基线平均降幅分别为1.57%和2.15%,均优于安慰剂组(0.14%)(p<0.0001)。玛仕度肽头对头度拉糖肽治疗2型糖尿病的III期临床研究(DREAMS-2)也已达到研究终点。
数据来源:NextPharma®数据库-注册性临床
信达生物于2024年2月向NMPA提交玛仕度肽用于成人肥胖或超重患者的长期体重控制的上市申请(CXHS2400010、CXHS2400009、CXHS2400008),且于2024年8月提交第二个适应症2型糖尿病的上市申请(CXHS2400071、CXHS2400072、CXHS2400070),目前两项适应症的上市申请均处在审评阶段,预计可于2025Q2获批。
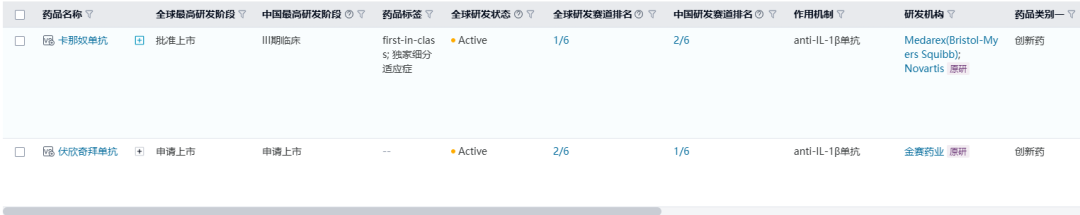
伏欣奇拜单抗(firsekibart)
预计批准时间:2025Q3
伏欣奇拜单抗是金赛药业研发的IL-1β单抗,2024年4月金赛药业向NMPA提交伏欣奇拜单抗用于治疗急性痛风性关节炎的上市申请, 截止目前,我国尚未有同作用机制药物上市。全球范围内,同靶点的仅有诺华的卡那奴单抗获批上市用于包括痛风性关节炎、幼年特发性关节炎在内的一系列适应症的治疗。
数据来源:NextPharma®数据库-基础查询
2024年召开的美国风湿病学会(ACR)学术年会公布了伏欣奇拜单抗的III期临床研究(GUARD-1,CTR20223136)结果,对急性痛风性关节炎发作的患者,对比复方倍他米松(得宝松)治疗和伏欣奇拜单抗治疗的效果。伏欣奇拜单抗组在72小时急性痛风性关节炎疼痛缓解的VAS评分上非劣效于复方倍他米松(57.1 vs 53.8 mm),并且显著延迟了基线至首次急性痛风复发的时间(Not estimable vs 45d)且在48小时后伏欣奇拜单抗组疼痛VAS评分下降程度比复方倍他米松组趋势更明显。因此对NSAIDs/秋水仙碱禁忌、不耐受或缺乏疗效/不适合反复使用类固醇激素的成年痛风性关节炎急性发作的患者,伏欣奇拜单抗能有效缓解发作疼痛,用药12周后也能有效延缓痛风的急性复发,并且可维持至用药后24周。
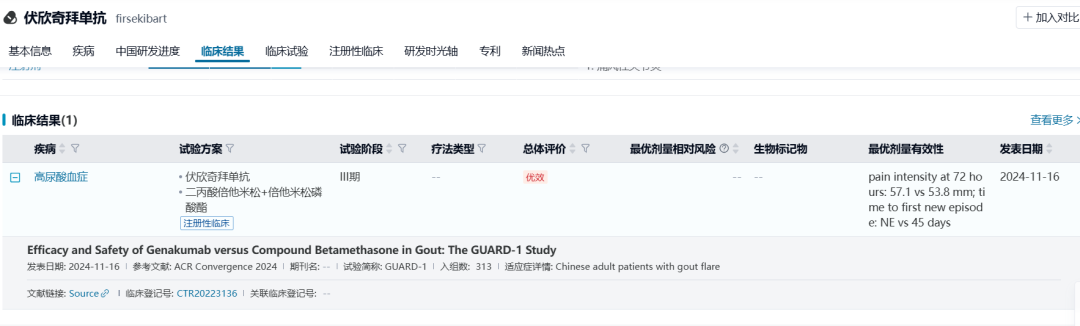
数据来源:NextPharma®数据库-临床结果
截止至2024年12月6日,除药理毒理外的药学/临床等已完成评审,伏欣奇拜单抗目前在CDE处于排队待审评阶段,预计于2025Q3获批。
罗莫索珠单抗(romosozumab,Evenity)
预计批准时间:2025Q4
罗莫索珠单抗是UCB和Amgen共同开发的anti-SOST单抗,通过特异性抑制骨硬化蛋白,提高骨形成并降低骨吸收从而降低骨折风险。
目前仅有罗莫索珠单抗1款作用于SOST的药物获批上市,于2019年在日本、美国和欧洲获批,用于治疗具有高骨折风险的骨质疏松症人群(包括绝经后妇女),但由于可能带来的心血管风险,FDA对罗莫索珠单抗给予黑框警告。2023年,罗莫索珠单抗全球销售额达11.6亿美元。
在中国,UCB和Amgen已完成一项3期临床研究(CTR20192682/NCT05067335),评价罗莫索珠单抗在中国绝经后骨质疏松症女性中的疗效、安全性和耐受性。研究结果显示, 327 名受试者在双盲期结束的第6个月时,罗莫索珠单抗组的lumbar spine BMD(腰椎骨密度)相对于基线的平均百分比变化显著高于安慰剂组(9.81% vs 0.44%),且两组之间的不良事件和严重不良事件保持平衡。
同机制的药品中,罗莫索珠单抗是国内进展最快的,于2024年7月首次向NMPA递交治疗骨质疏松症的上市申请(JXSS2400057),目前处在排队待审评阶段,预计于2025Q4获批。

数据来源:NextPharma®数据库-全球研发赛道排名
替利珠单抗(teplizumab,Tzield)
预计批准时间:2025Q4
替利珠单抗是全球首个且唯一一个可用于自身免疫性1型糖尿病的疾病修饰疗法,为1型糖尿病的预防和治疗提供了新的希望。替利珠单抗是一种不结合Fc受体的靶向CD3的单抗,通过与效应T细胞表面的CD3结合,抑制其对胰岛β细胞的攻击。替利珠单抗的Fc段经氨基酸修饰后可减少其与补体和Fc受体的结合,从而降低相关毒性反应。
礼来于2007年以4100万美元首付款从MacroGenics获得了替利珠单抗的独家权益,但在2010年因III期试验未达主要终点,礼来放弃进一步探索。2018年,Provention Bio从MacroGenics手中收购了替利珠单抗。1年后,替利珠单抗取得里程碑式进展并被FDA授予突破性疗法资格。2020年,Provention Bio启动替利珠单抗的滚动BLA,并于同年10月与赛诺菲达成合作,为替利珠单抗的商业化铺路。
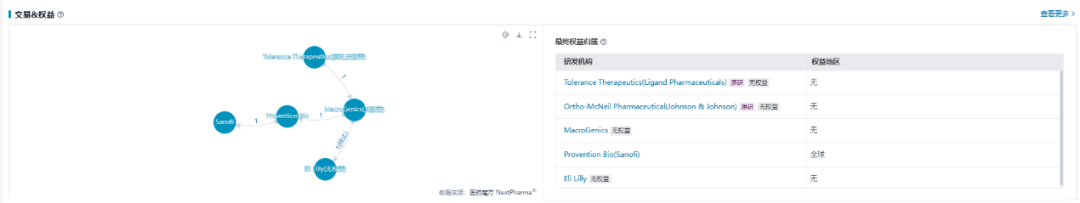
数据来源:NextPharma®数据库-医药交易
替利珠单抗于2022年11月在美国获批用于成人和8岁及以上儿童1型糖尿病2期患者,以延缓3期1型糖尿病发病,成为全球首个且唯一一个获FDA批准用于延缓1型糖尿病发病的免疫疗法。2023年,赛诺菲以29亿美元收购Provention Bio公司以获得替利珠单抗资产。
Provention Bio于2024年8月向NMPA提交替利珠单抗用于1型糖尿病的上市申请(JXSS2400070),并被纳入优先审评审批程序,或将成为我国首个1型糖尿病延缓发病治疗药物,预计2025Q4获批。
三、罕见病领域
索乐匹尼布(sovleplenib)
预计批准时间:2025Q2
索乐匹尼布是由和黄医药自主研发的一款口服选择性Syk抑制剂,在研适应症包括免疫性血小板减少症(ITP)、温抗体型自身免疫溶血性贫血(WAIHA)等。2022年1月,索乐匹尼布获得CDE授予的突破性疗法认定,用于治疗原发性免疫性血小板减少症。
免疫性血小板减少症是一种由血小板数目减少引起的出血性疾病,免疫系统会针对患者自身的血小板产生抗体并破坏它们,从而出现皮肤瘀斑、止血困难等症状。
2023年8月,和黄医药宣布索乐匹尼布治疗原发免疫性血小板减少症的ESLIM-01中国III期研究达到主要终点。ESLIM-01是一项索乐匹尼布在中国开展的随机、双盲、安慰剂对照的III期临床试验,共纳入188名既往接受过至少一种治疗的慢性成人原发免疫性血小板减少症患者。结果显示,接受索乐匹尼布治疗的患者的持续应答率取得了具有临床意义和统计学意义的显著改善(索乐匹尼布vs安慰剂:48.4% vs 0%, p<0.0001)
数据来源:NextPharma®数据库-基础查询
目前国内尚无Syk抑制剂获上市,基于ESLIM-01 III期研究结果,和黄医药于2024年1月在中国提交了索乐匹尼布的上市申请,用于治疗原发免疫性血小板减少症,预计于2025Q2获批,有望成为中国首款上市的Syk抑制剂。
BBM-H901
预计批准时间:2025Q3
BBM-H901是信念医药自主研发的一款针对factor IX的基因疗法,以静脉给药的方式将人凝血因子IX基因导入B型血友病患者体内持续表达,提高并长期维持患者的凝血因子水平,用于预防患者出血。
2022年,BBM-H901获得FDA孤儿药认定,同年又被CDE纳入突破性治疗品种名单。2023年10月,武田与信念医药宣布达成独家合作协议,获得BBM-H901注射液在中国内地、中国香港和中国澳门的商业化经营许可。
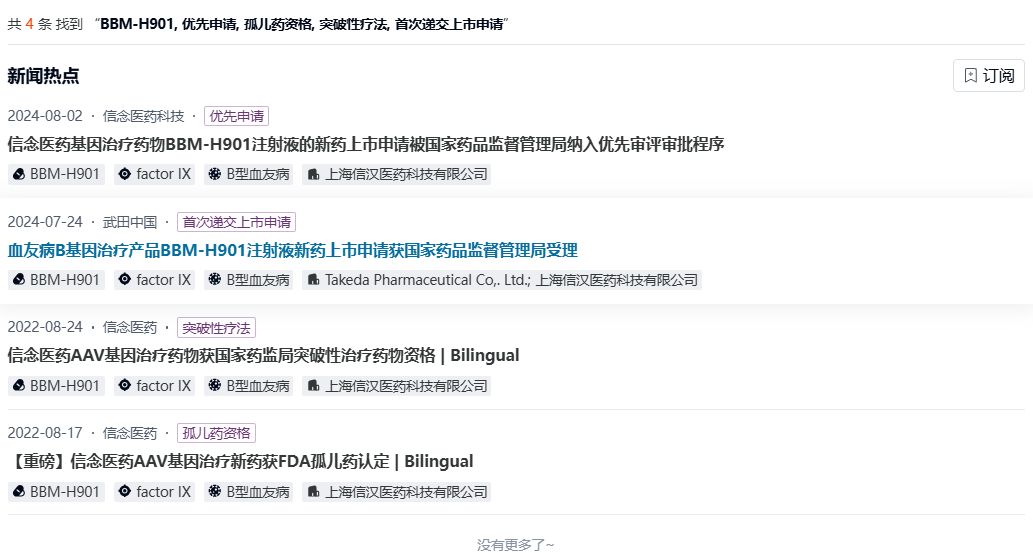
数据来源:NextPharma®数据库-新闻热点
信念医药与武田于2024年7月向CDE递交BBM-H901用于治疗B型血友病成年患者的上市申请,预计于2025Q3获批。本次递交是基于一项多中心、单臂、开放、单次给药注册临床研究(CTR20212816),旨在评估单次静脉输注BBM-H901注射液在≥18岁且内源性凝血因子Ⅸ(FⅨ)活性≤2 IU/dL(即≤2%)的B型血友病患者中的安全性和有效性。该研究的最新数据于2024 ASH年会上公布,BBM-H901显著降低年化出血率并提高FIX活性水平,且具有良好的安全性和耐受性。试验涉及26名参与者,80.8%的患者在治疗后未出现出血事件。
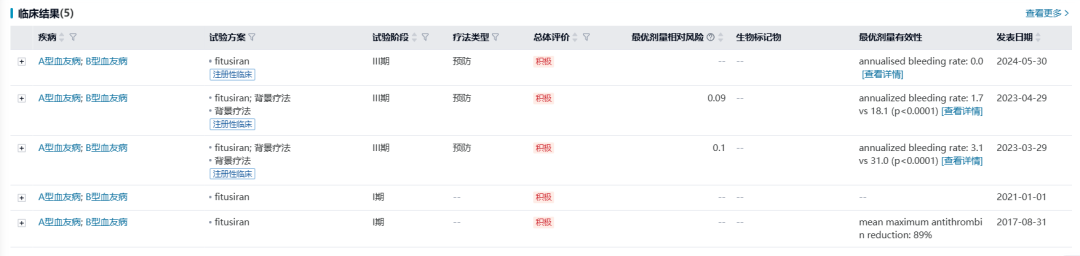
fitusiran
预计批准时间:2025Q3
fitusiran是赛诺菲研发的一款靶向抗凝血酶III(AT III)的siRNA疗法,旨在通过促进凝血酶的产生,用于有或无凝血因子VIII或IX抑制性抗体的A或B型血友病患者的常规预防治疗。fitusiran利用Alnylam Pharmaceuticals的 ESC-GalNAc技术平台,可实现每月一针皮下注射给药。
fitusiran的III期临床研究(ATLAS-A/B)显示,与对照组相比,每月一次皮下注射fitusiran预防治疗组患者的年化出血率降低了90% (95% CI [84.1%; 93.6%], P <0.0001) ,所有类型血友病人群均有显著临床获益。Fitusiran于2023年12月被FDA授予突破性疗法资格。此后,赛诺菲于2024年6月向FDA提交fitusiran的上市申请。
数据来源:NextPharma®数据库-临床结果
Fitusiran作为血友病领域的潜在first-in-class品种,已被CDE纳入《以患者为中心的罕见疾病药物研发试点工作计划(“关爱计划”)》试点项目。2024年5月,fitusiran的上市申请(JXHS2400033、JXHS2400034)获NMPA受理,目前处于审评阶段,预计2025Q3获批。
四、其他领域
Andexanet alfa(Andexxa,Ondexxya)
预计批准时间:2025Q2
Andexanet alfa是由Portola Pharmaceuticals开发的重组人凝血因子Xa(factor Xa,FXa)蛋白,2013年获FDA突破性疗法认定,最早于2018年5月在美国获加速批准上市,2019年4月和2022年3月又分别于欧洲和日本上市,是全球第一个也是唯一一个针对FXa抑制剂的解毒剂。
阿哌沙班、利伐沙班等FXa抑制剂用于预防和治疗血栓栓塞,尽管安全性和有效性优于华法林,但临床上缺乏能迅速逆转其抗凝作用的解毒剂,以降低治疗相关出血风险、满足这些患者的紧急手术需求。
Andexanet alfa是一种重组修饰的人FXa诱饵蛋白,无催化活性,在活性位点保留了能以高亲和力结合FXa抑制剂的能力(1:1结合模式)。Andexanet alfa在血管内结合并中和FXa抑制剂,从而恢复内源性FXa活性。凝血活性和血管栓塞就像天平的两端,Andexanet alfa能逆转抗凝作用,同时在治疗中也存在诱发动静脉血栓、缺血性事件等不良反应的风险,因此FDA对andexanet alfa给予黑框警告,EMA亦对致血栓风险做出警告说明。
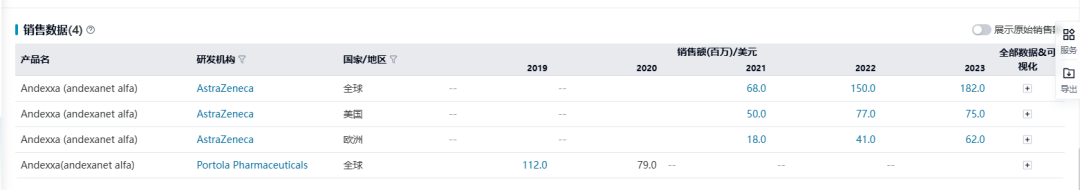
2020年7月,Alexion Pharmaceuticals完成对Portola Pharmaceuticals的收购,后者成为Alexion全资子公司。次年7月,阿斯利康以390亿美元收购Alexion,将Andexxa®纳入旗下。自阿斯利康接手以来,Andexxa®的销售额持续增长,2023年全球销售额为1.82亿美元。
数据来源:NextPharma®数据库-销售数据
今年6月,一项上市后IV期ANNEXA-I研究(NCT03661528)因预先达到止血优效性标准而提前终止。该研究旨在评估andexanet alfa相比常规疗法,在口服阿哌沙班和利伐沙班等FXa抑制剂后15小时内出现急性颅内出血的患者中的疗效和安全性。在一项452例患者的中期疗效评估中,andexanet alfa相比常规疗法止血效果分别为67% vs 53.1% (p=0.003)。基于此,阿斯利康将继续在美国和欧盟进行申报,以寻求andexanet alfa在这些地区的完全批准。
2023年12月16日,andexanet alfa注射剂首次在中国递交上市申请,目前处于排队待审状态,预计2025Q2在中国获批。
利生奇珠单抗(risankizumab, Skyrizi)
预计批准时间:2025Q2
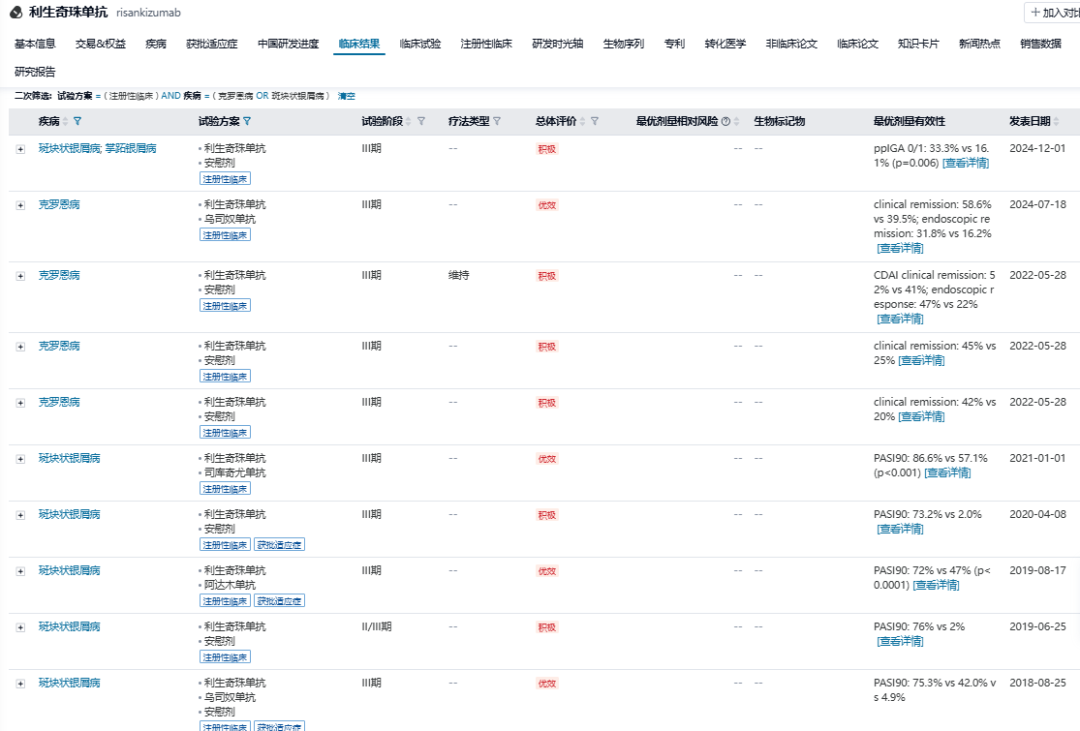
利生奇珠单抗是由勃林格殷格翰开发的一款IL-23p19单抗,Abbvie于2016年以5.95亿美元的首付款获得该产品的全球权益。2019年,利生奇珠单抗首次进入全球市场,目前已有斑块状银屑病、银屑病关节炎、克罗恩病、溃疡性结肠炎等多项适应症获批。2023年,利生奇珠单抗的销售额达到77.63亿美元,增速达50.3%,2024年销售额预计将突破110亿美元。
利生奇珠单抗针对斑块状银屑病、克罗恩病,已开展多项头对头注册性临床试验。2024年7月,NEJM发表了利生奇珠单抗vs乌司奴单抗治疗克罗恩病的临床结果,针对既往接受过生物制品治疗的克罗恩病患者,临床缓解率分别是:58.6% vs 39.5%,内镜缓解率分别是:31.8% vs 16.2%,利生奇珠单抗均取得优效性。
值得注意的是,与在国内已获批的anti-IL-23单抗不同,利生奇珠单抗在国内开展的适应症均为炎症性肠病,包括克罗恩病、溃疡性结肠炎。
数据来源:NextPharma®数据库-临床结果
2023年7月,利生奇珠单抗首次在华申报上市,推测申报适应症为克罗恩病,预计于2025年Q2获批。此外,2024年6月,利生奇珠单抗申报的新适应症上市申请获得CDE受理,推测申报适应症为溃疡性结肠炎。
莱博雷生(lemborexant, Dayvigo)
达利雷生(daridorexan,Quviviq)
预计批准时间:2025Q2 & 2025Q3
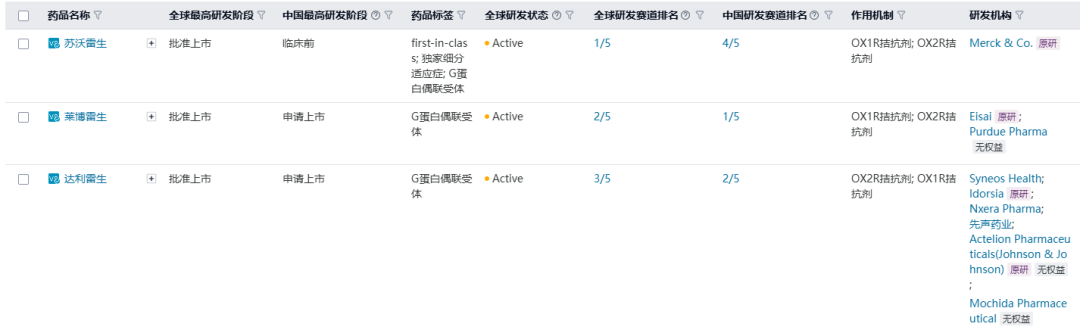
莱博雷生是Eisai研发的一款双重食欲素受体拮抗剂,通过竞争性结合两种食欲素受体亚型(OX1R和OX2R),抑制食欲素神经传递,调节睡眠-觉醒节律。根据中国成人失眠诊断与治疗指南(2023版),失眠的药物治疗主要包括苯二氮䓬受体激动剂 、双食欲素受体拮抗剂、褪黑素和褪黑素受体激动剂等。
目前全球已上市的双食欲素受体拮抗剂有苏沃雷生、莱博雷生、达利雷生,其中莱博雷生在中国进度最快,于2024年1月向CDE递交上市申请,达利雷生进度次之,于2024年7月在中国申请上市。
数据来源:NextPharma®数据库-基础检索
与传统用药方案相比,双食欲素受体拮抗剂具有不改变睡眠结构,没有次日残留效应和药物依赖性等优势。莱博雷生的一项注册性3期临床研究(NCT02783729)显示,莱博雷生vs唑吡坦vs安慰剂,持续睡眠的潜伏期:-19.5 vs -12.6 vs -6.5 min,显著改善了失眠患者的睡眠发作和睡眠维持,包括在后半晚,且耐受性良好。预计于2025Q2在中国获批。
达利雷生是先声药业从 Idorsia 公司引进的一款双食欲素受体拮抗剂,2024 年 5 月底,先声药业宣布达利雷生用于治疗中国失眠患者的 III 期临床试验(NCT06010693))达到主要终点。研究共纳入 206 例患者,主要研究终点为失眠患者夜间觉醒的减少时间,临床数据显示,在治疗的第 1 个月及第 3 个月,达利雷生较安慰剂显著改善了患者入睡、睡眠维持及自我报告的总睡眠时间,且不改变睡眠结构。此外,研究中未发现依赖性、反跳性失眠、戒断症状和药物滥用证据,显示良好的安全耐受性,且支持长期用药。目前在CDE处于排队待审评阶段,预计于2025Q3获批。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
