
Cancer Res:AFDN缺乏促进转移性结直肠癌的肝嗜性
来源:生物谷原创 2024-08-26 12:36
该研究表明AFDN缺乏可促进结直肠癌的肝向性转移,为研究结直肠癌肝转移机制提供了新的视角和认识。
癌细胞选择性转移到特定器官是一个复杂的过程,不仅受解剖学因素的影响,还受生物和器官特异性微环境因素的影响。转移前生态位是指原发肿瘤部位细胞产生的可溶性因子和细胞外囊泡(EVs),它们可以改变远处器官的微环境,以容纳迁移的癌细胞并促进其生长。此外,弥散性肿瘤细胞(dtc)可以在循环和新组织环境中进入休眠状态,逃避免疫监视,并与组织微环境相互作用,从休眠状态中唤醒。最后,转移性定植需要多种生物学过程,这些过程依赖于癌细胞的固有特性,以及靶器官细胞提供的允许的肿瘤微环境。
在结直肠癌肝转移过程中,结直肠癌细胞的固有特性与微环境中分子和细胞的相互作用都会影响转移。一旦CRC细胞成功进入肝脏微环境,它们就会与肝窦内皮细胞上的细胞粘附分子相互作用,导致弥散性肿瘤细胞(dtc)的包裹和粘附。
当大量肿瘤细胞侵入肝脏时,库普弗细胞(KCs)的抗肿瘤功能可能被抑制。结直肠癌细胞可能与KCs相互作用,刺激肝细胞生长因子(HGF)、血管内皮生长因子(VEGF)等生长因子的分泌,这种刺激促进肿瘤细胞增殖和血管生成。肝细胞在肝转移中也起着重要作用,肝细胞还可释放多种生长因子,均可促进肿瘤生长、运动和侵袭。然而,肝细胞促进肿瘤转移的机制尚不完全清楚。
AFDN位于染色体上,是粘附连接(AJs)的主要成分,它在邻近上皮细胞之间形成粘附连接。AFDN作为粘附连接的支架蛋白,可以募集cadherin或连接蛋白,并与actin相互作用,促进粘附连接的形成。AFDN在肿瘤中的作用首次在急性髓性白血病中被报道,在急性髓性白血病中,与赖氨酸特异性甲基转移酶2A (MLL)融合导致AFDN亚细胞定位改变和Ras信号通路激活,促进急性髓性白血病的进展,导致预后不良。然而,AFDN主要在各种实体肿瘤中起肿瘤抑制作用。
AFDN表达缺失或下调与结直肠癌、骨肉瘤、子宫内膜癌、乳腺癌和胰腺癌的不良预后相关。在胰腺癌中,AFDN的缺失不仅会破坏粘附和紧密连接,还会上调Snail的表达,促进上皮-间质转化(EMT)和转移。AFDN还通过与CRC中的囊性纤维化跨膜传导调节因子(CFTR)相互作用,维持黏着连接,抑制肿瘤细胞迁移和侵袭。然而,AFDN在结直肠癌患者肝脏中的生物学功能和分子机制尚不清楚。

图片来源:https://pubmed.ncbi.nlm.nih.gov/39047222/
近日,来自浙江大学医学院附属第四医院的研究者们在Cancer Res杂志上发表了题为“AFDN deficiency promotes liver tropism of metastatic colorectal cancer”的文章,该研究表明AFDN缺乏可促进结直肠癌的肝向性转移,为研究结直肠癌肝转移机制提供了新的视角和认识。
肝转移是结直肠癌患者发病和死亡的主要原因,更好地了解结直肠癌肝脏嗜性和转移的生物学机制有助于确定更好的预防和治疗策略。
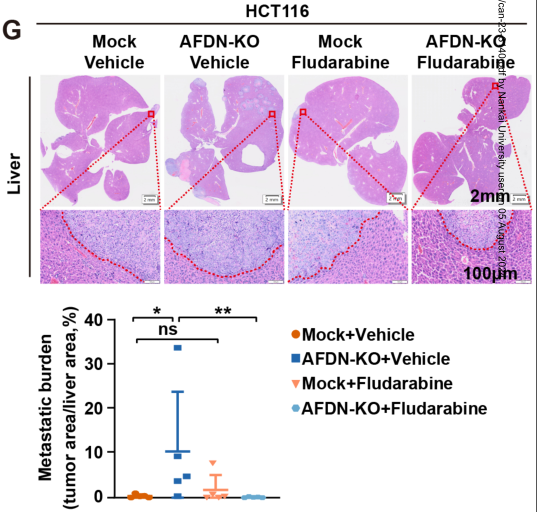
在这项研究中,研究者在小鼠结直肠癌模型中进行了基因组侧CRISPR功能缺失筛查,发现AFDN缺乏,AFDN是一种参与建立和维持细胞间接触的蛋白质,是肝转移的驱动因素。AFDN表达升高与结直肠癌患者生存期延长相关。缺乏afdn的结直肠癌细胞优先转移到肝脏,而不是肺。结直肠癌细胞原发部位的AFDN缺失通过破坏紧密的细胞间连接促进了癌细胞的迁移和侵袭。
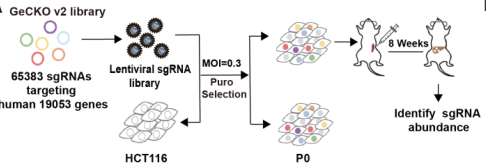
使用CRISPR/Cas9文库筛选结直肠癌肝转移相关基因的过程示意图
图片来源:https://pubmed.ncbi.nlm.nih.gov/39047222/
此外,通过JAK-STA T信号通路,CXCR4在afdn缺失的结直肠癌细胞中表达增加,从而降低afdn缺失的结直肠癌细胞的运动性,促进其在肝脏的定植。
CXCR4的表达受JAK-STAT信号的调控
图片来源:https://pubmed.ncbi.nlm.nih.gov/39047222/
综上所述,AFDN在癌症进展中的作用是非常复杂的,AFDN缺乏减少细胞间TJs,从而促进结直肠癌细胞的迁移和侵袭。AFDN缺乏也会抑制它们在肝脏中的运动,最终导致肝转移灶的形成,然而,AFDN缺陷在结直肠癌转移中特异性和肝向性的分子机制仍有待阐明。本研究为研究结直肠癌肝转移机制提供了新的视角和认识;然而,还需要进一步的研究。(生物谷 Bioon.com)
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
