
智力出现障碍的罪魁祸首!Cell Rep:两种“染色质管家”联手失控,神经元“身份混乱”致认知受损
来源:生物谷原创 2025-09-15 13:54
KDM1A 和 KDM5C 就像 “染色质管家”,联手维持神经元的 “身份认同”;一旦两者同时 “失灵”,神经元会 “乱表达基因”、染色质结构紊乱,最终导致严重的认知障碍。
智力障碍(ID)影响着全球约 2% 的人口,其中 50% 的病例与遗传因素直接相关。近年来,科学家发现,两种名为 KDM1A 和 KDM5C 的组蛋白去甲基化酶突变,是多种智力障碍的 “罪魁祸首”——KDM1A 突变会导致 “腭裂-精神运动迟缓-特殊面容综合征”(CPRF,OMIM#616728),KDM1A 则与 “Claes-Jensen X 连锁智力障碍”(MRXSCJ,OMIM#300534)密切相关。这些疾病不仅表现为认知低下,还常伴随发育异常,但其背后的神经元层面机制一直未被完全揭开。
近日,西班牙米格尔・埃尔南德斯大学与西班牙国家研究委员会联合团队在《Cell Reports》发表重磅研究,首次揭示:KDM1A 和 KDM5C 就像 “染色质管家”,联手维持神经元的 “身份认同”;一旦两者同时 “失灵”,神经元会 “乱表达基因”、染色质结构紊乱,最终导致严重的认知障碍。

KDM1A 和 KDM5C 虽同属组蛋白去甲基化酶,却分工互补:KDM1A 负责去除组蛋白 H3 第 4 位赖氨酸的单甲基化(H3K4me1)和双甲基化(H3K4me2),KDM5C 则专注于去除 H3K4me2 和三甲基化(H3K4me3);两者协同作用时,能将 “活跃基因标记” H3K4me3 逐步转化为 “沉默标记” H3K4me0,从而抑制非神经元基因的表达,确保神经元专注执行自身功能。
研究通过染色质免疫沉淀测序(ChIP-seq)发现,在成年小鼠海马神经元中,78% 的 KDM5C 结合基因同时也是 KDM1A 的靶点,这些共同靶点主要集中在基因启动子和增强子区域——启动子是基因 “开关”,增强子调控基因表达强度,两者共同守护神经元的转录稳态。
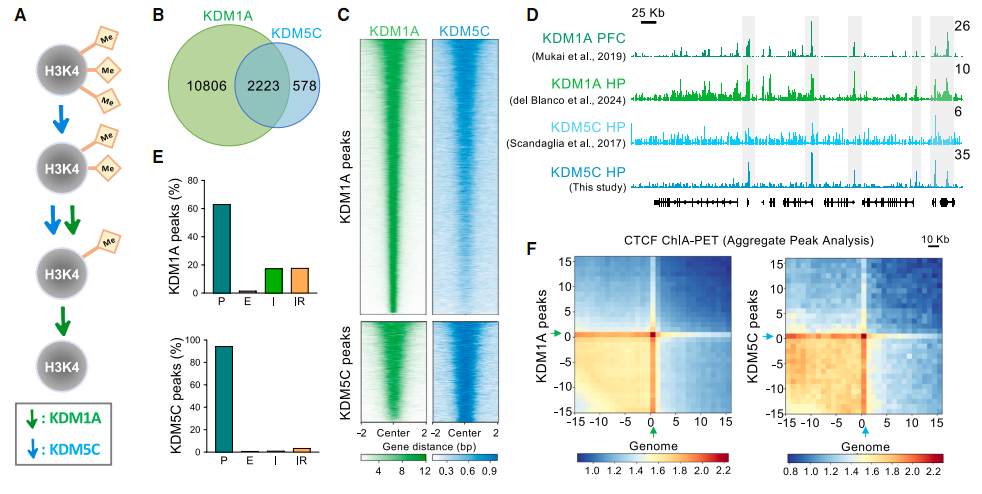
KDM1A和KDM5C在成年人神经元的基因组中有许多共同的靶点
为验证它们的协同作用,研究团队构建了“前额叶特异性双重诱导敲除小鼠”(dKDM-ifKO):通过 CamKIIα-creERT2 驱动系统,在小鼠 2 月龄时注射他莫昔芬(TMX),特异性敲除前额叶兴奋性神经元中的 Kdm1a 和 Kdm5c 基因,1 个月后分析分子和行为变化。
结果显示,这种双重敲除引发的异常远超单一基因敲除:转录组测序(RNA-seq)发现 408 个差异表达基因,其中 399 个显著上调,75% 是非神经元基因(如上皮细胞分化相关的 Tnxb、肾脏离子通道基因 Kcna7),这些基因本应在神经元中 “沉默”,却因两种酶缺失被异常激活;更关键的是,ChIP-seq 检测到 5388 个 H3K4me3 差异甲基化区域(DMRs),83% 是 “新生峰”——这些区域在正常神经元中完全没有 H3K4me3 标记,却在双重敲除后异常出现,意味着染色质的 “沉默状态” 被彻底打破。
进一步观察染色质结构发现,双重敲除还破坏了核内染色质的 “分区秩序”。通过超分辨显微镜,研究人员看到,正常神经元核内的 H3K27me3(另一种抑制性标记)会形成密集斑点,而 dKDM-ifKO 小鼠的 H3K27me3 斑点变得弥散,尤其在染色中心周围扩散——这种结构紊乱比单一敲除小鼠出现得更早(4 月龄即明显,单敲除需到老年才出现),说明 KDM1A 和 KDM5C 共同维持染色质的空间分区,一旦缺失,神经元的表观遗传架构会加速崩塌。
这些分子层面的异常,最终转化为严重的认知和行为问题。行为测试显示,4 月龄 dKDM-ifKO 小鼠在 “新物体位置识别(NOL)测试” 中无法区分物体的新旧位置,证明空间记忆受损;在 Morris 水迷宫中,它们找到隐藏平台的潜伏期显著延长,反转学习阶段(平台换位置)表现更差,且更倾向于贴着池壁游动(触觉趋性增加),提示认知灵活性下降。到 12 月龄时,小鼠还出现焦虑加剧——旷场测试中更多时间待在周边区域、明暗箱测试中回避光亮区域,探索行为减少,但运动协调能力(转棒测试)和痛觉敏感性(热板测试)未受影响,排除了运动障碍的干扰。
电生理实验进一步揭示了认知受损的神经元基础:dKDM-ifKO 小鼠海马 CA1 区锥体神经元的动作电位(AP)发放频率显著升高,后超极化(AHP)幅度降低——AHP 是 AP 后的电位抑制过程,能调节神经元兴奋性,其减弱会让神经元 “过度活跃”。这一变化与离子通道基因的异常表达直接相关:双重敲除后,非神经元离子通道(如 Otop1-3)被异位激活,而神经元特异性通道表达下降,打破了兴奋性平衡。
研究还发现,KDM1A 和 KDM5C 并非孤立作用,它们还与组蛋白甲基转移酶(KMTs,如 KMT2A、SETD1B)形成调控网络。对比 KMTs 条件敲除小鼠的数据集发现,KDM1A/KDM5C 与 KMT2A、SETD1B 共同调控的差异甲基化区域(DMRs),富集了突触可塑性、学习记忆、认知调控等生物学过程相关基因。这意味着,这些表观遗传因子联手守护认知相关基因的正常表达,任何一环失灵都可能引发智力障碍。
这项研究不仅揭开了智力障碍的关键机制——KDM1A 和 KDM5C 的协同作用是神经元 “身份维持” 的核心,还为治疗提供了新方向:未来可通过调控 H3K4 甲基化水平(如靶向补充去甲基化酶活性)或修复染色质空间结构,恢复神经元的正常基因表达,从而改善认知功能。当然,研究也存在局限,比如目前仅在小鼠模型中验证,未来还需在人类神经元模型中进一步确认,但这一发现已为理解智力障碍的表观遗传基础迈出了关键一步。(生物谷Bioon.com)
参考文献:
Ana M. Martín-González,Juan Paraíso-Luna,Sergio Niñerola, et al. Cooperative control of neuron-specific repressive chromatin states by intellectual-disability-linked KDM1A and KDM5C demethylases, Cell Reports (2025). DOI:10.1016/j.celrep.2025.116201
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
