
Nature Methods | 复旦大学原致远/中国科学院赵屹合作为空间聚类方法提供了一个全面的评估框架
来源:生物探索 2024-03-20 10:37
该研究比较分析发现了当前方法的几个局限性,包括识别小区域的挑战,缺乏多切片分析能力和大规模可扩展性问题。
复旦大学原致远及中国科学院计算技术研究所赵屹共同通讯在Nature Methods在线发表题为“Benchmarking spatial clustering methods with spatially resolved transcriptomics data”的研究论文,该研究利用空间解析转录组学数据对空间聚类方法进行基准测试。该研究对34个SRT数据(7个数据集)的13种计算方法进行了基准研究。基于准确性、空间连续性、标记基因检测、可扩展性和鲁棒性对其性能进行了评估。
该研究发现现有的方法在性能和功能方面是互补的,为给定的场景选择合适的方法提供了指导。在测试另外22个具有挑战性的数据集时,发现了识别非连续空间域的挑战和现有方法的局限性,突出了它们在处理最近的大规模任务方面的不足。此外,通过145个模拟数据,检验了这些方法对四种不同因素的鲁棒性,并评估了预处理和后处理方法的影响。该研究对现有的SRT数据空间聚类方法进行了全面的评估,为这个快速发展的领域的未来发展铺平了道路。

空间分辨转录组学(SRT)的进步使基因表达的多重空间定位成为可能,允许研究人员超越细胞聚类,通过提供额外的空间信息来识别高阶组织结构或空间域。通过空间聚类识别空间域已成为构建空间图谱的标准初始步骤,并已被证明在可视化组织解剖、推断组织空间连续性、检测区域特异性标记基因、挖掘发育和疾病的空间特征以及识别区域依赖的分子调控网络中至关重要。
尽管近年来基于概率图形模型和图神经网络(GNNs)的计算方法可用于识别空间域,但所使用的数据集和指标缺乏一致性和全面性,这构成了重大挑战。这些困难来自于空间技术的快速发展,某些应用中使用的有限的评估指标,以及依赖于由某些实验室生成的从特定技术和组织获得的数据集。尽管已经对空间解析转录组学数据,特别是与细胞类型相关的转录组学数据进行了基准研究,但仍然需要专门针对用于识别空间域的空间聚类方法进行全面的基准研究。
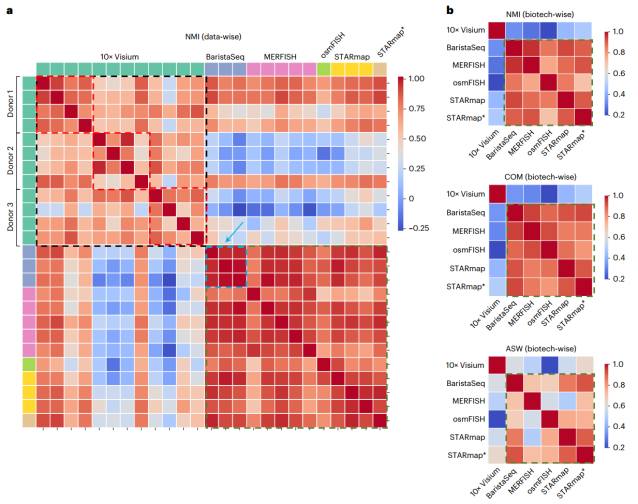
关于方法性能的数据之间的相关性(Credit: Nature Methods)
该研究对13种利用不同空间技术的空间转录组学数据识别空间域的方法进行了基准分析。研究人员从准确性、连续性、标记分数和可扩展性等方面对这些方法进行了评价,为空间聚类评估提供了一个全面的框架,可以帮助研究人员为其空间转录组学数据选择最佳的空间聚类工具。研究结果表明,没有一种方法在所有数据集上都是普遍有效的,最优方法取决于数据的特征。
该研究比较分析发现了当前方法的几个局限性,包括识别小区域的挑战,缺乏多切片分析能力和大规模可扩展性问题。随着空间转录组学数据对内存和计算时间的要求越来越高,需要更大的可扩展性来满足这些需求。对于一些确定的局限性,该研究提出了“分而治之”的方法来证明基准研究可以激励方法生物信息学家结合现有工具,以有效应对大规模空间数据集带来的挑战。总体而言,这项工作代表了对空间解析转录组学领域的深远贡献,为空间聚类方法提供了一个全面的评估框架,并促进了它们在各种数据集中的应用。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
