
Nature Methods | 全面评估长读长RNA测序:揭示转录组分析新前沿
来源:生物探索 2024-06-12 09:19
LRGASP项目通过系统评估多种长读长RNA测序技术和分析工具,提供了对当前最佳实践的深入理解和未来改进的方向。
长读长RNA测序技术(long-read RNA-seq technologies, lrRNA-seq)的兴起引发了对其全面评估的需求。为了评估长读长方法在转录组分析中的有效性,长读长RNA测序基因组注释评估项目(Long-read RNA-Seq Genome Annotation Assessment Project, LRGASP)应运而生。
该项目通过使用不同的测序协议和平台,生成了超过4.27亿条来自人类、小鼠和海牛物种的长读长序列数据。这些数据集包括从互补DNA(cDNA)和直接RNA数据集中提取的长读长序列,旨在解决转录本异构体(transcript isoform)检测、定量和新转录本检测中的挑战。(6月7日 Nature Methods “Systematic assessment of long-read RNA-seq methods for transcript identification and quantification”)
该研究表明,包含更长、更准确序列的文库能比那些仅增加读长深度的文库生成更准确的转录本,而增加读长深度则有助于提高定量准确性。在注释良好的基因组中,以参考序列为基础的工具展示了最佳性能。在使用无参考方法(reference-free approaches)或检测稀有和新型转录本时,建议结合额外的正交数据(orthogonal data)和重复样本。
LRGASP项目采用开放的社区合作模式,仿照之前成功的基准测试项目,测试了各种工具和平台在三个关键领域的表现:重建完整转录本、定量转录本丰度以及对缺乏高质量参考的基因组进行de novo转录本重建。长读长测序技术展示了其捕获完整和新型转录本的潜力,即使在熟知的基因组中也是如此。然而,不同生物信息学工具之间的一致性较低,反映了分析目标的差异。有效地定量转录本依然具有挑战性,长读长工具由于通量和错误限制,仍然落后于短读长工具。
该合作研究为当前的实践提供了一个基准,并为未来转录组分析方法的发展指明了方向。通过对人类、小鼠和海牛物种的RNA样本进行生物学三重重复实验,并在一个地点提取RNA后,分配给所有参与组进行测序,确保了数据的可靠性。综合使用cDNA制备方法和直接RNA测序,结合多种平台的数据,LRGASP项目全面评估了各种实验方法和分析工具在不同挑战中的表现,提供了对当前实践的深入洞察和未来改进的建议。
该研究的重要性在于它不仅为研究人员提供了长读长RNA测序技术在转录组分析中的实际应用指南,还为开发更高效的转录本检测和定量方法提供了有价值的参考。这些努力将有助于提高转录组数据的精度和可靠性,从而推动基因组学研究的进步。

近年来,随着长读长RNA测序技术(long-read RNA-seq technologies, lrRNA-seq)的迅速发展,对其在转录组分析中的应用前景引发了广泛关注。长读长RNA测序不仅能够提供更完整的转录本信息,还能够解决短读长测序(short-read sequencing)无法有效处理的复杂转录本异构体(transcript isoform)问题。然而,不同的测序平台和分析工具在转录本检测和定量方面的表现各异,为了全面评估这些技术的有效性和局限性,长读长RNA测序基因组注释评估项目(Long-read RNA-Seq Genome Annotation Assessment Project, LRGASP)应运而生。
LRGASP项目的目的是通过系统评估各种长读长RNA测序技术和分析工具,提供对当前最佳实践的指导,并为未来的发展指明方向 。
在LRGASP研究中,为了全面评估长读长RNA测序技术在转录组分析中的应用,研究团队设计并实施了一系列系统的方法和过程。
样本选择与制备
研究使用了来自人类、小鼠和海牛的样本,具体包括:
人类样本:使用了人类诱导多能干细胞系(WTC11)和人类胚胎干细胞系(H1-hES),以及从H1-hES细胞衍生的确定内胚层(H1-DE)。
小鼠样本:使用了小鼠胚胎干细胞系(ES)。
海牛样本:使用了海牛全血转录组。
所有样本都进行了生物学三重重复,确保数据的可靠性。RNA提取在一个中心进行,并分配给不同的参与组进行测序。
数据生成与测序平台
研究团队生成了超过4.27亿条长读长序列数据,采用了多种测序平台和协议,包括:
Oxford Nanopore Technologies(ONT):使用了cDNA测序试剂盒(PCS110)和直接RNA测序(dRNA)。
Pacific Biosciences(PacBio):使用了cDNA测序和改进的R2C2方法,以提高序列准确性。
CapTrap:用于富集5′-端有帽的RNA。
这些多样化的测序平台和方法确保了数据的广泛性和代表性,使得评估结果更具参考价值。
数据评估与分析方法
为了全面评估不同测序平台和工具的表现,研究团队设置了三个核心挑战,并针对每个挑战设计了详细的评估指标:
挑战1:重建完整转录本
评估指标:包括全长匹配转录本(Full Splice Match, FSM)、部分匹配转录本(Incomplete Splice Match, ISM)、新组合转录本(Novel In Catalog, NIC)和新转录本(Novel Not in Catalog, NNC)。
方法:使用SQANTI3工具对转录本进行分类,并结合正交数据(如短读长测序数据)进行验证。
挑战2:定量转录本丰度
评估指标:包括重复样本间的一致性、一致性相关系数(Consistency Measure, CM)、分辨率(Resolution Entropy, RE)等。
方法:使用多种工具对转录本丰度进行定量,并通过比较不同工具和平台的结果来评估其准确性和一致性。
挑战3:de novo转录本重建
评估指标:包括新转录本检测的敏感性和精度。
方法:使用无参考方法对数据进行分析,并与已知的转录本数据库进行比较。
具体过程与步骤
数据生成:在实验开始阶段,所有RNA样本经过标准化处理后,分别在不同的平台上进行测序。为了确保数据的一致性,所有样本均在同一地点进行RNA提取和初步处理。
数据处理:生成的序列数据通过标准的生物信息学流程进行预处理,包括质量控制、读长过滤和错误校正等步骤。不同的工具和平台可能会采用不同的数据处理方法,以确保最终数据的准确性和完整性。
分析工具的选择与应用:研究团队邀请了14个工具和实验室提交其预测结果,这些工具包括Bambu、FLAIR、FLAMES、IsoQuant、TALON、LyRic等。每个工具对同一数据集进行了分析,并提交了其在三个挑战中的预测结果。
评估与验证:为了避免利益冲突,评估和验证工作由不参与预测提交的LRGASP组织者小组完成。评估方法包括使用SIRV(Spike-In RNA Variant)数据集进行基准测试、模拟数据和基因手动注释数据集进行验证等。
结果分析与比较:通过综合评估工具在不同挑战中的表现,研究团队分析了各工具和平台在转录本重建、定量和de novo重建中的优劣。结果显示,不同工具在处理不同类型数据时表现出显著差异,且某些工具在处理低丰度转录本时存在较大挑战。
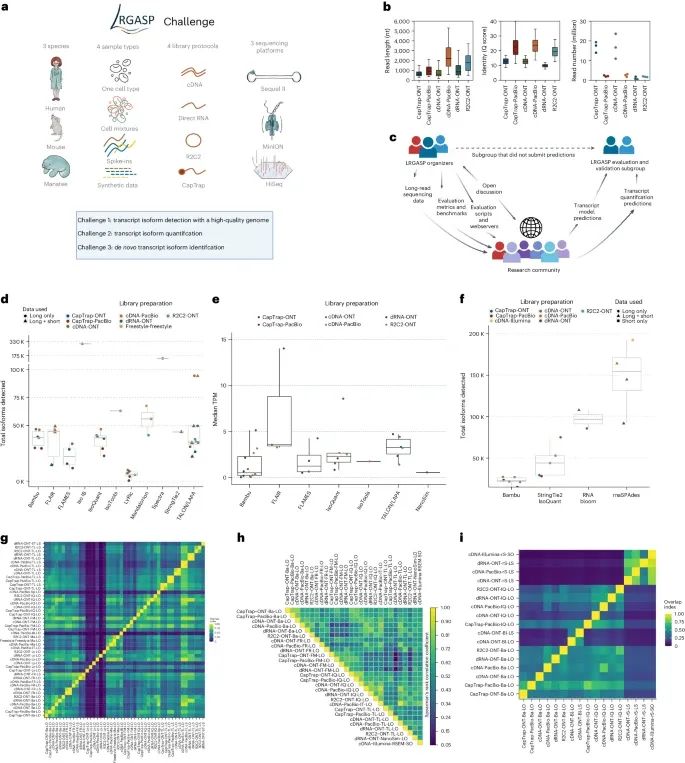
LRGASP(长读长RNA测序基因组注释评估项目)的概览,包括数据生成、读长分布、合作设计、各工具在不同数据类型上的表现,以及独特剪接链(UJC)的相对重叠情况(Credit: Nature Methods)
a. LRGASP项目的数据生成
用于LRGASP项目的数据生成情况。研究团队使用多种文库制备方法和测序平台,包括:CapTrap-ONT(Oxford Nanopore Technologies),CapTrap-PacBio(Pacific Biosciences),cDNA-ONT,cDNA-PacBio,dRNA-ONT,R2C2-ONT
这些方法生成了超过4.27亿条长读长序列数据,这些数据来自人类、小鼠和海牛物种。这些数据用于评估在转录本重建、丰度定量和de novo转录本检测中的表现。
b.读长分布、质量评分和测序深度
WTC11样本中读长分布、质量评分(Q score)和测序深度(每个生物重复)的分布情况。不同文库制备方法生成的读长分布和质量评分有所不同。例如,CapTrap-ONT和cDNA-ONT方法生成了约10倍于其他方法的读数。
c.LRGASP的合作设计
LRGASP项目的合作设计。项目邀请了工具开发者提交对三大挑战的预测,包括转录本异构体检测、转录本丰度定量和de novo转录本重建。为了避免利益冲突,评估和验证工作由未提交预测的LRGASP组织者小组完成。整个LRGASP研究设计和评估基准通过公开设计进行,以确保公平和透明的基准测试。
d. 挑战1:转录本异构体检测
各工具在挑战1(转录本异构体检测)中对人类WTC11样本的不同数据类型上报告的转录本数量。提交的工具包括Bambu、FLAIR、FLAMES、IsoQuant、TALON等。工具检测到的转录本数量差异显著,从几百到数十万个不等。
e. 挑战2:转录本丰度定量
各工具在挑战2(转录本丰度定量)中对人类WTC11样本的不同数据类型上报告的中位TPM值(Transcripts Per Million)。提交的工具包括IsoQuant、FLAIR、Bambu等。不同工具在定量低丰度转录本时表现较差,且丰度相关性较低。
f. 挑战3:de novo转录本重建
各工具在挑战3(de novo转录本重建)中对小鼠ES细胞数据的不同数据类型上报告的转录本数量。提交的工具包括FLAMES、TALON、rnaSPAdes等。工具在检测新转录本时的敏感性和精度差异较大。
g. 独特剪接链(UJC)的相对重叠
各提交中报告的独特剪接链(UJCs)的成对相对重叠情况。每个提交的UJCs作为每行的参考集合。图中显示了来自列提交的UJCs的重叠分数,以热图形式展示。例如,一个提交如果包含了许多其他提交的UJCs子集,其行中的重叠分数将较高,但列中的分数则较低。数据仅显示了WTC11的提交结果。
h. TPM值的Spearman相关性
挑战2中各提交的TPM值之间的Spearman相关性。不同工具在转录本丰度定量上的相关性较低,表明在丰度定量方法上存在显著差异。
i. UJC的成对相对重叠
进一步展示了UJCs在每个提交中的成对相对重叠情况。与图1g类似,图中显示了每个提交的UJCs作为参考集合时,来自列提交的UJCs的重叠分数。
文库质量对转录本重建的影响
研究表明,包含更长和更准确序列的文库在转录本重建中表现更好。具体来说,在注释良好的基因组中,基于参考序列的工具表现最佳。例如,研究中使用的Bambu、FLAIR、FLAMES和IsoQuant工具在检测全剪接匹配转录本(Full Splice Match, FSM)时表现出高精度,而TALON、IsoTools和LyRic则检测到较多的不完全剪接匹配转录本(Incomplete Splice Match, ISM)。值得注意的是,LyRic在分析中未使用现有注释,这解释了其检测结果。
在具体数据上,不同的方法在检测已知基因和转录本时表现出显著差异。例如,使用不同方法检测到的已知基因数量从399到23,647不等,而检测到的转录本数量从524到329,131不等。大多数工具报告了每个基因平均3-4个转录本,但Spectra和Iso_IB工具报告了大量的转录本(分别约170,000和330,000)。
转录本定量的挑战
在定量转录本丰度时,研究发现增加读长深度有助于提高定量的准确性。然而,大多数工具在定量低丰度转录本时表现较差。例如,在细胞混合实验中,不同工具在预期和观察到的丰度之间的相关性(SCC)范围为0.74至0.87,除Bambu工具外,RSEM工具在细胞混合实验中的表现最佳,SCC为0.87,MRD为0.13,NRMSE为0.38。
在SIRV-Set 4数据和模拟数据中,RSEM工具同样表现出色,其平均SCC为0.84,MRD为0.12,NRMSE为0.45,而其他长读长工具的表现则有所不及。例如,NanoSim的SCC为0.78,MRD为0.23,NRMSE为0.89,IsoQuant的SCC为0.76,MRD为0.19,NRMSE为0.89。
新转录本检测的敏感性和精度
对于新转录本的检测,研究中所有工具的敏感性都较低,且精度差异较大。例如,Bambu工具在检测新转录本时的NNC和NIC值最低,而IsoQuant和TALON工具的表现也相对较好。相比之下,FLAIR和Mandalorion工具检测到约20%的NIC转录本,但NNC百分比较低。
研究还发现,文库制备和测序平台的差异显著影响了转录组定义。例如,文库制备方法如CapTrap和cDNA测序生成了更多的读长数据,但不同工具对这些数据的处理结果不同。在具体实验中,不同平台生成的读长数量存在显著差异,这表明在选择测序平台时需要综合考虑文库质量和读长数量。
研究团队还设计了多种评估指标和方法,以便更全面地评估不同工具的表现。例如,使用SIRV数据集进行基准测试、模拟数据和基因手动注释数据集进行验证等。这些方法帮助揭示了不同工具在处理复杂转录组数据时的优势和不足。
LRGASP项目通过系统评估多种长读长RNA测序技术和分析工具,提供了对当前最佳实践的深入理解和未来改进的方向。这项研究的重要性不仅在于它为科学家提供了实际应用的指导,还在于它为未来长读长RNA测序技术的发展奠定了基础。通过不断改进和优化这些技术,我们将能够更好地理解基因表达和调控的复杂机制,从而推动生命科学和医学研究的前沿发展 。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
