
Cell Metabolism:顾伟团队报道磷脂酸过氧化介导的铁死亡在肿瘤抑制中的作用
来源:生物世界 2024-02-05 13:26
该研究揭示了由膜结合蛋白PHLDA2介导的一条酶催化的非经典的磷脂酸(phosphatidic acid,PA)过氧化诱导的铁死亡通路在肿瘤抑制中发挥关键作用。
铁死亡(Ferroptosis)是一种铁离子依赖的由过量脂质过氧化积累导致的细胞膜破裂的程序性细胞死亡。通过体内体外对铁死亡机制和功能的研究发现,靶向铁死亡是一种肿瘤精准治疗的潜在方法。然而直接靶向管家基因GPX4诱导的铁死亡会对正常组织产生不可避免地损伤:2022年11月,阿斯利康和宾夕法尼亚大学研究人员在 Nature 上发文证实经典铁死亡诱导剂会导致肿瘤内免疫细胞,特别是中性粒细胞的铁死亡,从而破坏肿瘤免疫从而促进肿瘤生长。因此,探索不依赖于经典铁死亡抑制剂的体内激活铁死亡来治疗肿瘤的方法是一个亟需解决的科学问题。
哥伦比亚大学欧文医学中心癌症医学研究所顾伟教授团队自2015年以来在铁死亡抑制肿瘤发生发展领域做出了一系列的工作:2015年在Nature发文揭示p53通过抑制胱氨酸转运蛋白SLC7A11表达促进铁死亡,建立了p53及p53乙酰化与铁死亡新的联系;2019年在Nature Cell Biology发文提出p53介导的铁死亡是通过脂质加氧酶ALOX12由高ROS驱动下实现的;2021年在Nature Communications发文揭示p53转录调控磷脂酶iPLA2β能够发挥独立于GPX4的调控铁死亡机制;2023年在Cell Metabolism发文报道p53可以通过抑制维生素K代谢促进铁死亡从而抑制肿瘤的发生发展。
2024年2月2日,顾伟教授团队(杨鑫博士为第一作者)再次在 Cell Metabolism 期刊上发表了题为:PHLDA2-mediated phosphatidic acid peroxidation triggers a distinct ferroptotic response during tumor suppression 的研究论文。
该研究揭示了由膜结合蛋白PHLDA2介导的一条酶催化的非经典的磷脂酸(phosphatidic acid,PA)过氧化诱导的铁死亡通路在肿瘤抑制中发挥关键作用。

首先,研究团队通过CRISPR-Cas9全基因组敲除筛选发现膜结合蛋白PHLDA2和经典铁死亡诱导基因ACSL4都可以作为关键调控因子参与胱氨酸饥饿导致的铁死亡。细胞内无论是PHLDA2或ACSL4的敲除均会降低胱氨酸饥饿后脂质过氧化的水平和铁死亡。由于胱氨酸饥饿不仅会导致谷胱甘肽(GSH)水平下降从而抑制GPX4功能,还会导致活性氧(ROS)水平上调。因此,他们对细胞进行有机过氧化物处理,包括过氧化氢叔丁醇(TBH,tert-butyl hydroperoxide)或过氧化氢异丙苯(CMH,cumene hydroperoxide),来探索模拟高ROS水平的条件下细胞铁死亡的调节。研究团队首先证明8小时以内TBH或CMH处理导致的细胞死亡全部是铁死亡。
他们进一步发现PHLDA2敲除细胞显著抑制高水平ROS诱导的脂质过氧化的水平和细胞死亡,而ACSL4敲除不会改变细胞对高水平ROS诱导的铁死亡的敏感度。众所周知,ACSL4敲除会显著降低细胞对GPX4抑制剂(例如RSL-3)导致的铁死亡敏感度,但是PHLDA2敲除细胞对于GPX4抑制剂诱导的铁死亡没有影响。这些实验说明PHLDA2是胱氨酸饥饿或高水平ROS诱导的铁死亡中不可缺少的因子,其功能独立于经典的ACSL4-GPX4介导的铁死亡通路。
顾伟课题组前期研究证明催化多不饱和脂肪酸(PUFA)氧化的脂氧合酶ALOX12是高水平ROS诱导的铁死亡中的关键因子,但是严格定位于细胞质内的ALOX12如何发挥促进膜上脂质过氧化的机制仍不明确。通过对纯化的ALOX12结合蛋白质复合物进行质谱分析,研究团队鉴定出PHLDA2是ALOX12的结合蛋白。通过一系列体内体外生物化学实验和细胞铁死亡实验,他们证明ALOX12与PHLDA2结合对于高水平ROS诱导的铁死亡至关重要。
考虑到ACSL4/GPX4通路调控的铁死亡的机制主要是由磷脂酰乙酰胺(PE, phosphatidylethanolamine)过氧化所决定的,研究团队进一步通过体外靶向磷脂氧化实验探索PHLDA2- ALOX12介导的高水平ROS诱导的铁死亡的机制。他们首先检测PHLDA2 与不同类型磷脂之间的结合亲和力,发现PHLDA2和PA具有显著的结合能力,且与PE不结合。尽管ALOX12本身不能够结合任何磷脂,但在PHLDA2存在时ALOX12也能够显著结合PA,且不结合PE,说明PHLDA2能招募ALOX12至特定种类的磷脂。进一步通过靶向氧化磷脂组学,作者发现PHLDA2能显著增强ALOX12催化的花生四烯酸酯化磷脂(18:0/20:4-PA)的过氧化,且抑制18:0/20:4-PE的过氧化,说明PHLDA2通过其选择性结合磷脂的能力调控ALOX12对于磷脂的选择性过氧化。进一步,研究团队在ACSL4/GPX4双敲除细胞中孵育含有18:0/20:4-PA的脂质体,此时过表达PHLDA2和ALOX12能够显著增强细胞铁死亡的水平,证实PHLDA2/ALOX12复合物可以通过催化的带有长链不饱和脂肪酸的PA的过氧化在没有经典铁死亡诱导剂的条件下启动铁死亡。
为了进一步阐明PA介导的高水平ROS诱导的铁死亡的内在代谢调控机制,研究团队比较了RSL-3和TBH处理的CRISPR-Cas9全基因组敲除筛选结果。他们发现GPAT3是PA合成酶中唯一一个在TBH诱导的铁死亡起到关键作用的。GPAT3敲除细胞显著抑制胱氨酸饥饿或TBH诱导的铁死亡,而在RSL-3诱导的铁死亡中没有明显作用。通过磷脂代谢组学分析,他们发现TBH处理后可以显著降低PUFA-PA的含量且敲除GPAT3或者铁死亡抑制剂Ferrostatin-1处理可以完全挽救这个现象,证明高水平ROS诱导的铁死亡中存在PUFA-PA的消耗。进一步通过氧化磷脂代谢组学分析,他们发现所有类型的磷脂在TBH处理后都会出现过氧化水平上升的现象,但是只有PUFA-PA的氧化会被敲除GPAT3或者铁死亡抑制剂Ferrostatin-1处理完全挽救。很有意思的是,敲除GPAT3的细胞在TBH处理下并没有出现细胞死亡现象,但是PUFA-PE的过氧化仍然维持在很高的水平,说明PUFA-PE的过氧化在高水平ROS诱导的铁死亡中的作用微乎其微。
最后,为了阐明PHLDA2介导的PA过氧化诱导的铁死亡在体内肿瘤抑制中的作用,作者首先利用免疫缺陷的异种移植模型探索PHLDA2在肿瘤生长中的作用。和ACSL4敲除组相比,PHLDA2敲除显著促进肿瘤的生长。通过各项铁死亡标志物的分析,研究团队发现与ACSL4敲除组相比,PHLDA2敲除的肿瘤中表现出明显的铁死亡抑制现象。他们进一步在免疫系统完整的小鼠中构建了致癌物(DEN,diethylnitrosamine)诱发的肝癌模型和Eµ-Myc诱导的淋巴瘤模型,发现PHLDA2敲除会显著增强肿瘤的发生发展,证明PHLDA2介导的铁死亡在肿瘤抑制中发挥关键的作用。
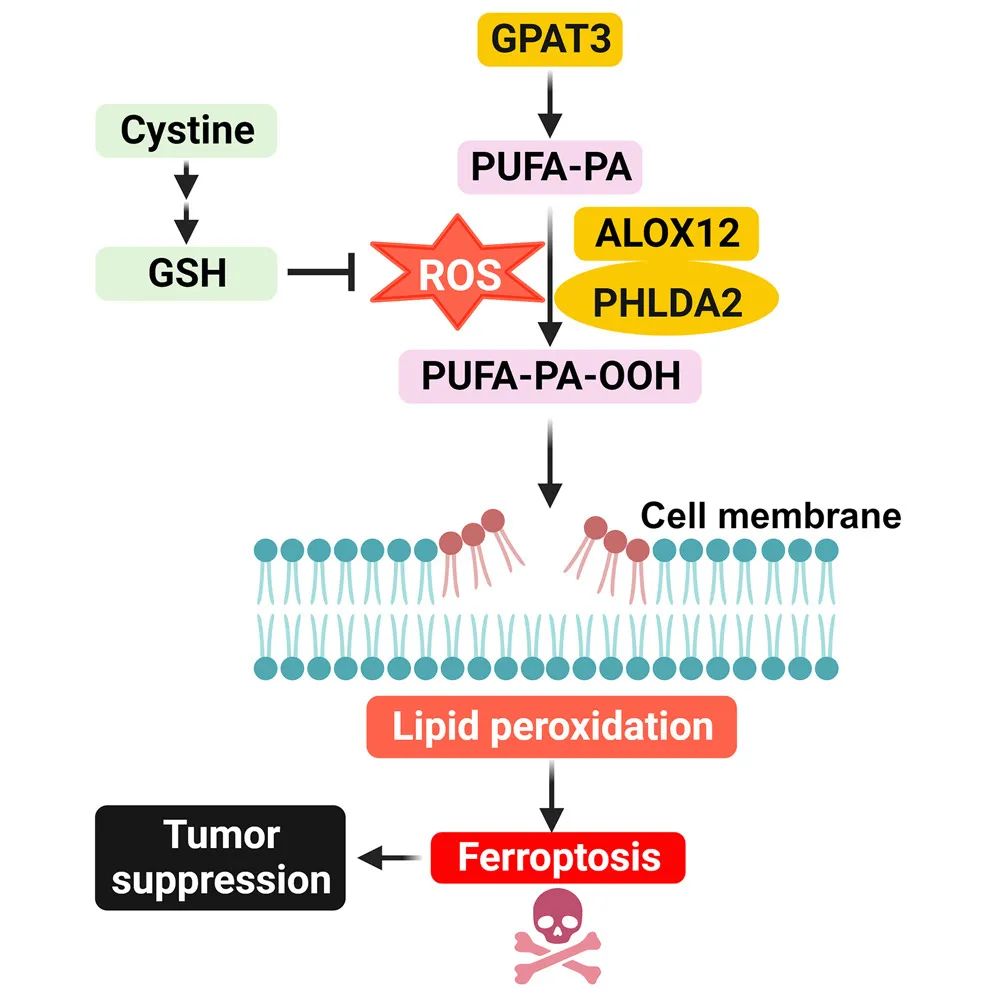
综上所述,该研究证明了PHLDA2介导的铁死亡对于机体抵抗肿瘤发生至关重要。考虑到PHLDA2介导的铁死亡不需要经典铁死亡诱导剂的诱导,而是在高水平ROS积累下自然发生的,这项工作将为肿瘤治疗中靶向铁死亡提供了新思路。
哥伦比亚大学顾伟教授为论文通讯作者,杨鑫博士为论文第一作者。该工作得到了匹兹堡大学Valerian E. Kagan教授、哥伦比亚大学Hülya Bayir教授、Brent R. Stockwell教授和张志国教授以及纪念斯隆-凯特琳癌症中心姜学军教授等合作者的大力支持。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
